高效液相色譜-電感耦合等離子體質(zhì)譜法測(cè)定接骨七厘片中總砷含量及砷形態(tài)分析
2020-08-12 02:22:30錢(qián)保勇
中國(guó)藥業(yè) 2020年15期
朱 瓊,方 靜,蔡 鵬,錢(qián)保勇
(江蘇省泰州市食品藥品檢驗(yàn)所,江蘇 泰州 225300)
接骨七厘片是由醋乳香、醋沒(méi)藥、當(dāng)歸、土鱉蟲(chóng)、龍血竭、酒大黃等藥材加工制成的中成藥,具有活血化瘀、接骨止痛的功效[1],能促進(jìn)骨折愈合,增強(qiáng)抗折作用,對(duì)小鼠醋酸致痛有鎮(zhèn)痛作用,臨床應(yīng)用廣泛。但接骨七厘片中的有害元素時(shí)有超標(biāo)[1],特別是砷超標(biāo)需引起重視。為控制接骨七厘片的質(zhì)量,評(píng)價(jià)其安全性,砷的測(cè)定十分有必要。砷的毒性根據(jù)其不同形態(tài)有著顯著差異,亞砷酸鹽As(Ⅲ)和砷酸鹽As(Ⅴ)是毒性最大的無(wú)機(jī)砷,一甲基砷(MMA)和二甲基砷(DMA)的毒性較弱,而砷甜菜堿(AsB)和砷膽堿(AsC)無(wú)毒。為了科學(xué)地評(píng)估接骨七厘片中的砷毒性,確定其安全使用的含量范圍,有必要對(duì)砷形態(tài)進(jìn)行分析。目前,中藥中總砷的測(cè)定方法主要有氫化物原子熒光法、電感耦合等離子體質(zhì)譜法、電感耦合等離子體發(fā)射光譜法,對(duì)砷形態(tài)的分析方法主要有高效液相色譜-氫化物原子熒光法、高效液相色譜-電感耦合等離子體質(zhì)譜法(HPLC-ICP-MS)[2-3]。本研究中采用HPLC-ICPMS 法對(duì)中成藥中砷形態(tài)進(jìn)行了含量測(cè)定,并制訂了相應(yīng)質(zhì)量標(biāo)準(zhǔn),可為研究其他中成藥質(zhì)量標(biāo)準(zhǔn)提供參考。現(xiàn)報(bào)道如下。
1 儀器與試藥
1.1 儀器
Ultimate 3000 型高效液相色譜儀(美國(guó)Dionex 公司);ICP -Q 型電感耦合等離子體質(zhì)譜儀(美國(guó)賽默飛公司);梅特勒電子天平(美國(guó)梅特勒公司,精度為0.1 mg)。
1.2 試藥
砷單元素標(biāo)準(zhǔn)溶液(批號(hào)為175385-28,1000μg/mL),亞砷酸鹽As(Ⅲ)(批號(hào)為144014-7,1 000 μg/mL),砷酸鹽As(Ⅴ)(批號(hào)為976236-2,1 000 μg/mL),均購(gòu)自美國(guó)O2Si Smart Solutions 公司;砷甜菜堿[批號(hào)為1509,以砷計(jì)(38.8±1.1)μg/g],砷膽堿[批號(hào)為1512,以砷計(jì)(28.1±1.1)μg/g],一甲基砷[批號(hào)為16072,以砷計(jì)(25.1±0.8)μg/g],二甲基砷[批號(hào)為1607,以砷計(jì)(52.9±1.8)μg/g],均購(gòu)自中國(guó)計(jì)量科學(xué)研究院;接骨七厘片(湖南金沙藥業(yè)有限責(zé)任公司,批號(hào)分別為161235,161237,170120);硝酸為色譜純,水由Milli-Q系統(tǒng)制得;其他試劑均為分析純。
2 方法與結(jié)果
2.1 砷形態(tài)分析
2.1.1 HPLC-ICP 質(zhì)譜條件
質(zhì)譜條件:等離子體功率為1 550 W,載氣為高純氬氣;載氣流速為1.0 L/min;采樣深度為5 mm;冷卻氣流量為14 L/min;輔助氣流量為0.8 L/min;載氣流量為0.9 L/min;數(shù)據(jù)重復(fù)采集次數(shù)為3 次。
色譜條件:賽默飛AS7 陰離子交換柱(250 mm×4.6 mm);流動(dòng)相為10 mmol/L 碳酸銨(A),100 mmol/L碳酸銨(B),梯度洗脫,洗脫程序見(jiàn)表1;流速為0.8 mL/min;進(jìn)樣量為20 μL。記錄離子流圖(見(jiàn)圖1)。
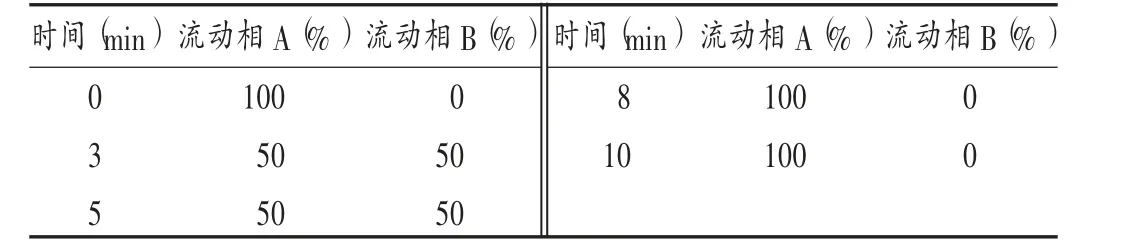
時(shí)間(min)流動(dòng)相B(%)流動(dòng)相A(%)流動(dòng)相B(%)時(shí)間(min)流動(dòng)相A(%)0 3 5 100 50 50 0 50 50 8 10 100 100 0 0
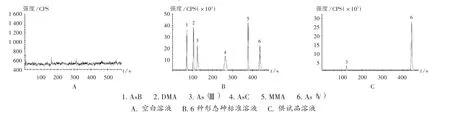
圖1 離子流圖
2.1.2 標(biāo)準(zhǔn)溶液配制及線性關(guān)系考察
精密量取砷單元素標(biāo)準(zhǔn)溶液適量,逐級(jí)稀釋配制成含砷為0,1.0,2.0,4.0,8.0,16.0,20.0 ng/mL 的系列質(zhì)量濃度混合標(biāo)準(zhǔn)溶液,照2.1.1 項(xiàng)下條件測(cè)定。取10個(gè)空白溶液測(cè)定空白強(qiáng)度值,按空白值的3 倍標(biāo)準(zhǔn)差和10 倍標(biāo)準(zhǔn)差分別計(jì)算檢測(cè)限(LOD)和定量限(LOQ)。
取6 種形態(tài)砷,精密稱定,分別用去離子水配制成As(Ⅲ)的質(zhì)量濃度分別為0,1.0,2.0,4.0,8.0,16.0,20.0 μg/L;As(V)的質(zhì)量濃度分別為0,1.0,2.0,4.0,8.0,16.0,20.0 μg/L;MMA 的 質(zhì) 量 濃 度 分 別 為0,1.00,2.00,4.00,8.01,16.02,20.02 μg/L;DMA 的質(zhì)量濃度分別為0,1.00,2.01,4.02,8.03,16.06,20.08 μg/L;AsB 的質(zhì)量濃度分別為0,1.02,2.04,4.08,8.15,16.30,20.38 μg/L;AsC 的質(zhì)量濃度分別為0,1.00,2.01,4.02,8.04,16.08,20.10 μg/L。按2.1.1 項(xiàng)下條件測(cè)定,標(biāo)準(zhǔn)曲線及相關(guān)系數(shù)見(jiàn)表2。取10 個(gè)空白溶液測(cè)定空白強(qiáng)度值,并計(jì)算LOD 和LOQ,結(jié)果見(jiàn)表2。
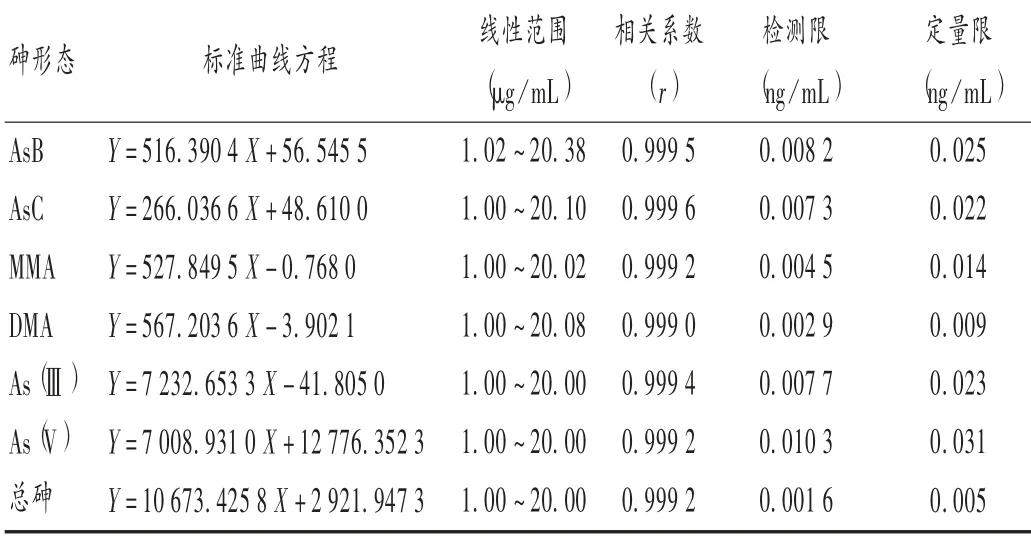
表2 標(biāo)準(zhǔn)曲線方程與靈敏度考察結(jié)果
2.2 總砷含量測(cè)定
2.2.1 質(zhì)譜條件
同2.1.1 項(xiàng)下質(zhì)譜條件。
2.2.2 溶液制備
取樣品(去包衣),粉碎,混勻,取粉末0.50 g,精密稱定,置100 mL 具塞錐形瓶中,加入人工腸液20 mL,超聲15 min,放置過(guò)夜。次日放入37.5 ℃烘箱中提取3 h,每隔0.5 h 振搖錐形瓶1 min。提取完畢,吸取中層濾液10 mL,用微孔濾膜過(guò)濾,取續(xù)濾液5 mL,再用0.02 mol/L乙二胺四醋酸二鈉(EDTA)溶液稀釋至50 mL 容量瓶中即得砷形態(tài)溶液。
取樣品(去包衣)0.25 g,精密稱定,置微波消解罐中,加入硝酸7 mL,加蓋密閉,在100 ℃下預(yù)消解15 min,冷卻,然后放入微波消解儀中消解。消解采用程序升溫,20 ℃升至110 ℃,保持8 min;110℃升至160 ℃,保持6 min;160 ℃升至180 ℃,保持25 min。待消解完全后,當(dāng)冷卻溫度至低于45 ℃時(shí),取出消解罐,繼續(xù)放冷至室溫,將消解后的溶液定量至50 mL 容量瓶中,用少量超純水潤(rùn)洗消解罐4 次,合并至容量瓶中,用超純水定容,搖勻,放置,取上清液作為總砷測(cè)定的供試品溶液。依法制備空白溶液。
2.2.3 方法學(xué)考察
精密度試驗(yàn):取樣品(批號(hào)為161235)粉末,分別加入高、中、低質(zhì)量濃度(16,8,2 ng/mL)的AsB、AsC、MMA、DMA、As(Ⅲ)、As(V)、總砷標(biāo)準(zhǔn)溶液,每個(gè)質(zhì)量濃度制備3 個(gè)平行樣,按擬訂方法測(cè)定,計(jì)算RSD。結(jié)果見(jiàn)表3,RSD在1.78% ~7.20%之間,精密度符合微量元素分析的要求。
穩(wěn)定性試驗(yàn):精密吸取同一供試品溶液,分別于0,1,2,4,8 h 時(shí)按擬訂條件測(cè)定。結(jié)果AsB,AsC,MMA,DMA,As(Ⅲ),As(V)和總砷離子強(qiáng)度的RSD均小于5.00% (n= 5),表明供試品溶液在8 h 內(nèi)穩(wěn)定。
重復(fù)性試驗(yàn):取樣品粉末(批號(hào)為161235),平行配制6 份砷形態(tài)樣品和6 份總砷樣品,按擬訂條件測(cè)定,根據(jù)砷形態(tài)測(cè)定結(jié)果計(jì)算的RSD為3.12% ~6.64%(n=6),根據(jù)總砷的測(cè)定結(jié)果計(jì)算的RSD為2.32%(n=6),符合微量元素分析要求。
加樣回收試驗(yàn):在已知含量的樣品(批號(hào)為161235)粉末中分別加入高、中、低質(zhì)量濃度(16,8,2 ng/mL)的AsB,AsC,MMA,DMA,As(Ⅲ),As(V)和總砷標(biāo)準(zhǔn)溶液,每個(gè)質(zhì)量濃度制備3 個(gè)平行樣,按擬訂條件測(cè)定,計(jì)算加樣回收率。結(jié)果見(jiàn)表4,其范圍在89.15% ~99.95%之間,RSD符合微量元素分析要求。
2.2.4 樣品含量測(cè)定
按擬訂條件測(cè)定3 批樣品中總砷和砷形態(tài)的含量,結(jié)果見(jiàn)表5。
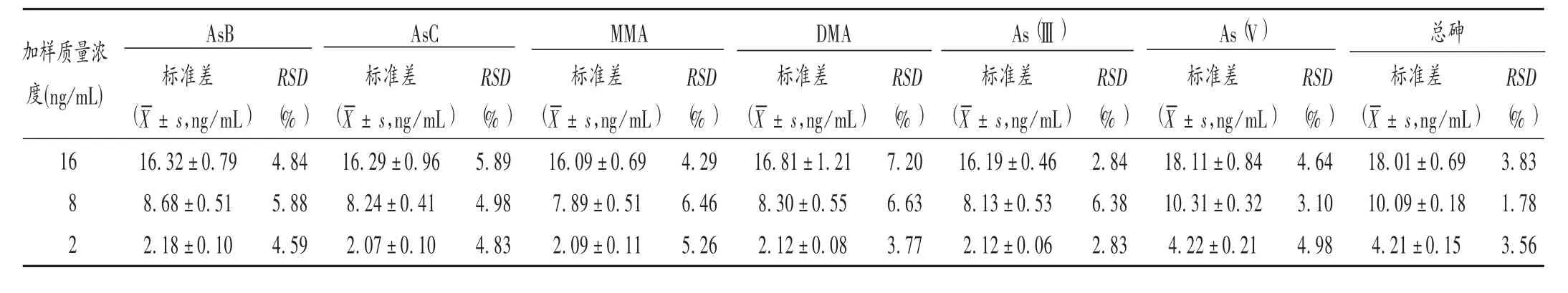
表3 AsB、AsC、MMA、DMA、As(Ⅲ)、As(V)、總砷精密度試驗(yàn)結(jié)果
3 討論
3.1 提取溫度選擇
比較人工胃液、人工腸液、水提液3 種提取方式在37.5 ℃的提取率,結(jié)果水提取和人工胃液提取的含量都很低,但人工腸液提取率略好。設(shè)置人工腸液提取的溫度分別為37.5,60,90,120℃,結(jié)果90℃提取效率最高,當(dāng)提取溫度繼續(xù)增加至120 ℃時(shí),As(Ⅲ)含量變低,As(Ⅴ)含量變高,提示砷形態(tài)會(huì)發(fā)生轉(zhuǎn)變。為了保證提取效率和砷形態(tài)不發(fā)生變化,故選擇人工腸液在90 ℃提取[4-6]。
表4 加樣回收試驗(yàn)結(jié)果(±s,%,n=3)
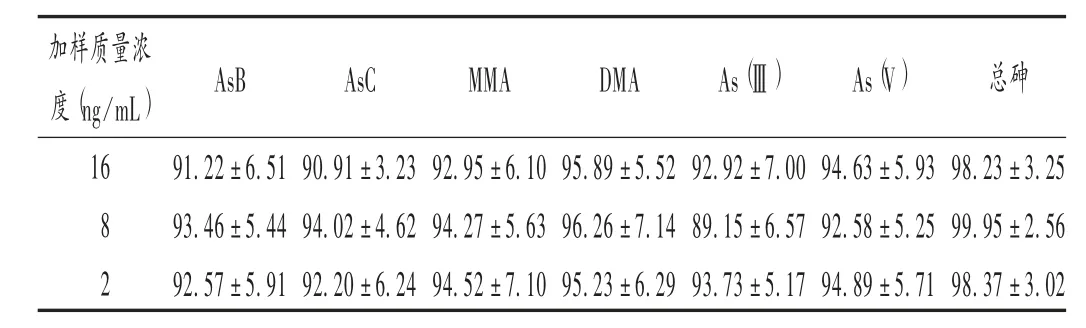
表4 加樣回收試驗(yàn)結(jié)果(±s,%,n=3)
加樣質(zhì)量濃度(ng/mL)AsB AsC MMA DMA As(Ⅲ)As(V)總砷16 8 2 91.22±6.51 93.46±5.44 92.57±5.91 90.91±3.23 94.02±4.62 92.20±6.24 92.95±6.10 94.27±5.63 94.52±7.10 95.89±5.52 96.26±7.14 95.23±6.29 92.92±7.00 89.15±6.57 93.73±5.17 94.63±5.93 92.58±5.25 94.89±5.71 98.23±3.25 99.95±2.56 98.37±3.02

表5 3 批樣品中砷形態(tài)測(cè)定結(jié)果(mg/kg,n=3)
3.2 流動(dòng)相選擇
2015年版《中國(guó)藥典(四部)》中砷形態(tài)測(cè)定采用的是0.025 mol/L 磷酸二氫銨(氨水調(diào)節(jié)pH 至8.0)為流動(dòng)相,本研究中采用的是碳酸銨為流動(dòng)相,是因?yàn)榱姿岫滗@更易污染質(zhì)譜的采樣錐,易導(dǎo)致質(zhì)譜熄火,而碳酸銨系統(tǒng)進(jìn)入ICP-MS 后,被分解成易揮發(fā)的氣體,不會(huì)產(chǎn)生殘留,可減少質(zhì)譜熄火等情況的發(fā)生。同時(shí),高鹽樣品經(jīng)過(guò)電感耦合等離子體時(shí)間太長(zhǎng),會(huì)產(chǎn)生信號(hào)漂移,應(yīng)以質(zhì)控樣品或?qū)φ杖芤哼M(jìn)行回測(cè),或采用內(nèi)標(biāo)法進(jìn)行校正。
3.3 溶劑選擇
對(duì)供試品進(jìn)行前處理時(shí),最后需將樣品用EDTA 溶液進(jìn)行稀釋定容,可有效降低溶劑效應(yīng),分離效果更好,其質(zhì)譜離子流圖也更滿意,特別是AsB 和MMA 的峰形會(huì)更尖銳,否則會(huì)出現(xiàn)雙峰及拖尾[7-12]。
3.4 分離方法選擇
目前,分離形態(tài)砷的方法主要有高效液相色譜-氫化物原子熒光法和HPLC-ICP-MS 法。前者分離時(shí)間較長(zhǎng),出峰時(shí)間短,峰形較差。HPLC-ICP-MS 法分離時(shí)間較短,且配制試劑較少,測(cè)定結(jié)果更穩(wěn)定,該方法可用于藥物體內(nèi)代謝途徑及部位的研究[13-14]。
3.5 方法評(píng)價(jià)
本研究中建立了測(cè)定接骨七厘片中砷形態(tài)的HPLC-ICP-MS 法,同時(shí)采用ICP-MS 法對(duì)接骨七厘片中的總砷進(jìn)行了測(cè)定,結(jié)果準(zhǔn)確、可靠。不同批次接骨七厘片中砷形態(tài)含量存在差異,其中As(V)含量最高,其次是As(Ⅲ),還含有少量的MMA,DMA,AsB 和AsC均未檢測(cè)到。由可溶性砷含量的測(cè)定結(jié)果可見(jiàn),接骨七厘片中的砷主要是可溶性砷,以可溶性砷中的As(V)含量最高。
