液相色譜—原子熒光聯用測定海水中無機砷和有機砷
2020-08-19 00:39:04李艷蘋王翠翠劉小騏
海洋技術學報 2020年3期
李艷蘋,王翠翠,劉小騏,高 艾
(自然資源部天津海水淡化與綜合利用研究所,天津 300192)
重金屬污染是危害最大的環境問題之一。砷是有毒且致癌的元素,對環境和人類健康都十分有害,是海洋環境污染監測的重要指標。但元素的有效性、毒性不能簡單地取決于其總濃度含量,而是在很大程度上取決于它的化學形態[1]。砷包括無機砷和有機砷,無機砷有三價砷As(Ⅲ)和五價砷As(Ⅴ),有機砷主要有一甲基砷(MMA)、二甲基砷(DMA)、砷甜菜堿(AsB)、砷膽堿(AsC)和砷糖。一般認為,無機砷的毒性很大,有機砷的MMA 和DMA 的毒性較小,AsB、AsC 和砷糖無毒[2-3]。也有報道稱,無機砷形態的毒性是有機砷的100 倍,三價砷的毒性是五價砷的60 倍[4]。砷元素的不同形態的差異比較大,因此砷形態的研究日益受到多個領域學者們的重視。
元素形態的分析方法有電化學分析法、光譜法、色譜法、質譜法等,隨著痕量元素形態分析技術的發展,高選擇性的分離技術與高靈敏的檢測技術聯用顯示出很大的應用優勢。目前在食品、醫藥、環境樣品等多個領域有很多砷形態的研究[1,5-13]。也有一些學者進行了自來水、河水、海水等水環境樣品的砷形態測定研究,如方懷防等[14]、Tupiti W 等[15]及Salaün P 等[16-17]等分別利用金電極伏安法測定了自來水、河水以及海水中無機砷形態,采用Na2SO3還原As(Ⅴ)為更有電活性的As(Ⅲ)來實現總砷的測定,然后通過總砷與As(Ⅲ)之差求出As(Ⅴ)的含量;有研究在更負的電勢條件下直接測定As(Ⅴ),無需還原為As(Ⅲ),在任何pH 條件均可測定As(Ⅲ)。伏安法有簡單、快速、成本低的優點,但一般只能進行無機砷形態的測定。氫化物發生技術可使離子態砷選擇性地還原為砷化氫或有機衍生物,與樣品基體相分離,能大大降低基體產生的干擾,近年來常與原子吸收光譜、原子熒光光譜、等離子體質譜等方法聯用進行砷的形態分析。Alp O 等[18]和張桂香[19]分別報道了原子熒光測定飲用水和海水中無機砷形態的方法,分別采用硫代乙醇酸和硫脲、抗壞血酸還原 As(Ⅴ)為 As(Ⅲ),As(Ⅲ)可直接測定,用差減法求得As(Ⅴ)含量。潘用樹等[20]用氫化物—原子吸收法測定了海水中的總無機砷、有機砷MMA 和 DMA,需要外加鋯測定 As(Ⅲ)等操作,但仍不能同時測出幾種砷形態。 Nakazato T 等[21]研究建立了氫化物結合液相色譜—電感耦合等離子體質譜(LC-ICP-MS)技術,用于海水中As(Ⅲ)、As(Ⅴ)及MMA 的測定,但需要配置氫化物發生系統,操作相對復雜,成本相對較高。行業標準《海水中三價砷和五價砷形態分析 原子熒光光譜法》(HY/T 152-2013)規定了測定海水中As(Ⅲ)和As(Ⅴ)的原子熒光光譜法,As(Ⅲ)與硼氫化鉀溶液反應生成氫化物進行測定,加入硫脲—抗壞血酸將As(Ⅴ)轉化為As(Ⅲ)后進行總砷的測定,As(Ⅴ)由總砷與 As(Ⅲ)差值得出,但該標準方法僅能測定海水中無機砷形態。
雖然目前開展了很多水環境樣品中砷形態的研究,但海水中砷形態的研究還比較少,測定方法還不完善,尤其是同時測定無機砷和有機砷形態的方法鮮有報道,因此建立海水中砷形態簡便且準確的測定方法有著重要的意義。不僅可為海水污染調查、海洋環境保護提供研究基礎和手段,還將為砷形態的遷移轉化及海水資源綜合利用提供重要參考[13,19]。本文基于液相色譜—原子熒光聯用技術的高選擇性、高靈敏性、使用成本較低等優點,結合砷形態的毒性及主要存在形式,主要進行了海水中三價砷、五價砷以及甲基砷形態的測定研究。
1 實驗與方法
1.1 儀器與試劑
儀器:LC-AFS6500 液相色譜—原子熒光形態分析儀(北京海光儀器有限公司);KQ3200B 超聲波清洗儀(昆山超聲儀器有限公司);Milli-Q 型超純水儀(美國 Millipore 公司);ST3100 型 pH 計(奧豪斯儀器(常州)有限公司);PAL-06S 型海水鹽度計(日本 ATAGO)。
試劑:鹽酸(優級純,天津風船化學試劑科技有限公司);氫氧化鉀(優級純,天津市津科精細化工研究所);硼氫化鉀(KBH4,優級純,東方化工廠);氯化鈉、氯化鉀、氯化鈣、六水合氯化鎂、七水合硫酸鎂、磷酸二氫鉀(KH2PO4)、十二水合磷酸氫二鈉(Na2HPO4·12H2O)(均為分析純,國藥集團化學試劑有限公司);亞砷酸鹽(GBW08666)、砷酸鹽(GBW08667)、一甲基砷(GBW08668)和二甲基砷(GBW08669)標準物質均購于國家標準物質研究中心,濃度分別為 75.7 μg/mL,17.5 μg/mL,25.1 μg/mL,52.9 μg/mL。砷單標及混合標準儲備溶液分別由上述標準物質配制,然后冷藏儲存,臨用時稀釋。
1.2 儀器操作條件
1.2.1 液相色譜條件及參數 液相泵模式:等度A泵;蠕動泵轉速:60 r/min;采樣頻率:10 Hz;流速:1.0 mL/min;進樣體積:100 μL。色譜柱:Hamilton PRP-X100 分析柱(250 mm× 4.1 mm,10 μm);柱溫:25 ℃;流動相:稱取0.610 4 g 十二水合磷酸氫二鈉、2.063 g 磷酸二氫鉀放入500 mL 燒杯中,用約200 mL 高純水溶解,完全溶解后再用高純水稀釋至刻度,搖勻,然后用0.45 μm 微孔濾膜過濾,再經超聲脫氣后方可使用。
1.2.2 原子熒光條件及參數 砷空心陰極燈(北京有色金屬研究院);砷燈主電流:60 mA;砷燈輔電流:30 mA;載氣流量:300 mL/min;屏蔽氣流量:900 mL/min;負高壓:300 V;載流:5% HCl;還原劑:0.5%KOH,2% KBH4。
1.3 樣品采集與處理
采樣器皿為聚乙烯材質,使用前器皿均在硝酸溶液(1+3)浸泡24 h 以上,并用超純水洗凈。海水樣品用 0.45 μm 微孔纖維濾膜過濾,加酸至 pH<2,密封冷藏。
2 測量數據與實驗結果
2.1 流動相pH 的影響
流動相pH 的不同,會影響砷形態的出峰、保留時間及分離效果。流動相的不同pH 值對砷形態保留時間的影響如圖1 所示。
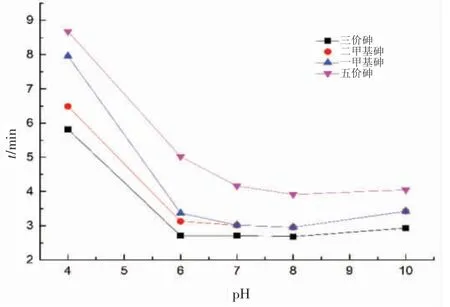
圖1 流動相pH 對砷形態保留時間的影響
從圖1 可以看出,隨著pH 逐漸增大至7.0 時,三價砷、五價砷、一甲基砷及二甲基砷4 種形態的保留時間大大縮短,一甲基砷與二甲基砷的保留時間越來越近;當pH 從7.0 增大至10.0 時,三價砷與五價砷的保留時間變化不大,但是兩種甲基砷的保留時間相吻合,二者色譜峰重疊。綜合考慮保留時間、分離效果、流動相pH 值,以及與無機砷相比,水環境樣品中甲基砷含量更低的情況,可選定流動相pH 為7.0,進行三價砷、甲基砷(一甲基砷與二甲基砷總量)、五價砷的分離,即可較好地解決海水中無機砷和有機砷的測定問題。
2.2 流動相濃度的影響
采用十二水合磷酸氫二鈉和磷酸二氫鉀配制流動相,分別配制成含磷酸二氫鉀不同濃度的流動相:5 mmol/L,10 mmol/L,15 mmol/L,22 mmol/L,30 mmol/L,在流動相 pH 為 7.0,流速為 1.0 mL/min 條件下,考察流動相的不同濃度對砷形態保留時間的影響,結果如圖2 所示。

圖2 流動相濃度對砷形態保留時間的影響
從圖2 可以看出,隨著流動相濃度逐漸增大,一甲基砷與二甲基砷兩種甲基砷的保留時間越來越近至相吻合,同時甲基砷的保留時間與三價砷、五價砷的保留時間也越來越接近。當流動相濃度為10 mmol/L 時,兩種甲基砷出峰部分重疊,出現多峰且拖尾嚴重。當流動相濃度在22~30 mmol/L 時,三價砷與甲基砷保留時間越來越接近,兩峰不能完全分離。當流動相濃度為22 mmol/L,減小流速至0.4 mL/min 時,三價砷才能與甲基砷完全分離。當流動相濃度為15 mmol/L 時,三價砷、甲基砷(一甲基砷與二甲基砷總量)及五價砷有較好的分離效果,因此選定流動相濃度為15 mmol/L。
2.3 不同流速的影響
對實際海水加標樣品,進行了流速對三價砷、甲基砷以及五價砷的影響研究,如圖3 所示。
當流速在0.4~1.2 mL/min 范圍內,隨著流速的不斷增大,三價砷和甲基砷的保留時間逐漸變小,且二者的保留時間越來越相近,而五價砷的保留時間則大大減小,后略微增大。從保留時間和分離效果綜合考慮,流速可選擇為0.8~1.0 mL/min。
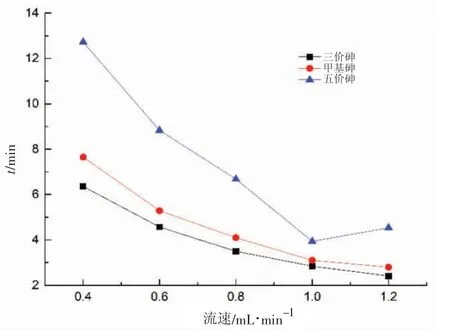
圖3 流速對砷形態保留時間的影響
2.4 鹽度的影響
為研究海水及濃鹽水基體鹽度對砷形態測定的影響,配制了砷形態濃度相同,鹽度分別為20,30,40,50,60 的鹽溶液。其中濃鹽水的配制如下:稱取氯化鈉46.94 g、氯化鉀1.30 g、氯化鈣1.95 g、六水合氯化鎂8.71 g、七水合硫酸鎂11.88 g,溶于少量純水中,定容至1 000 mL,混勻,相當于每千克海水中的含鹽量為60 g,不同鹽度的溶液可由此稀釋配制,并用鹽度計測定。
實驗表明,隨著鹽度的逐漸升高,三價砷的保留時間基本不變,甲基砷與五價砷的保留時間逐漸變小;當鹽度在40~60 范圍內,五價砷的靈敏度隨著鹽度的升高而下降,峰形變寬,甲基砷與三價砷的峰出現部分重疊,二者分離效果變差,鹽度對砷形態分離的影響應該是Cl-引起的。有文獻研究表明,海水中的 Na+,K+,Ca2+,Mg2+,Mn2+,A13+,Cr3+,Pb2+,Zn2+和Cu2+,均對測定結果無影響,而海水中存在的大量Cl-對砷形態的分離有干擾,使三價砷、一甲基砷及二甲基砷不能完全分離[22]。Cl-在經過陰離子交換色譜柱時,占據了大多離子交換的點位,從而影響砷形態在離子交換柱上的分離。綜合考慮靈敏度以及分離效果,測定海水及濃鹽水中三價砷、甲基砷以及無機砷時,可將樣品適當稀釋降低鹽度至20~30范圍內,采用基體匹配法測定以消除鹽度的影響。
2.5 工作曲線及精密度
采用鹽度為30 的人工海水配制砷形態混合標準系列溶液,濃度分別為 2 μg/L,5 μg/L,10 μg/L,20 μg/L,40 μg/L,50 μg/L,80 μg/L。在上述實驗優化的檢測條件下,以各濃度色譜峰面積對應標準溶液濃度繪制工作曲線,得到回歸方程。砷形態的線性方程、相關系數和檢出限見表1。
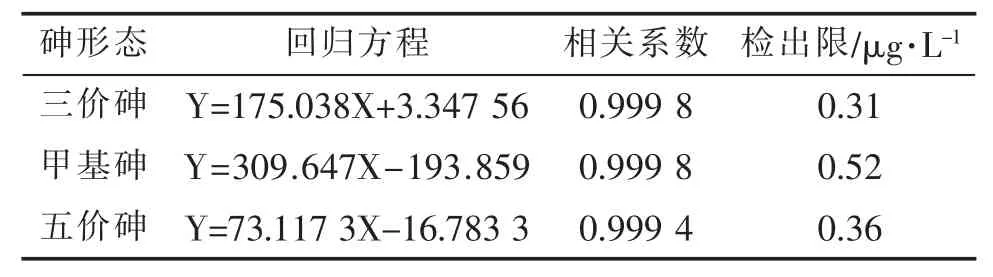
表1 砷形態的線性方程、相關系數和檢出限
由表1 可知,在優化的條件下,在2~80 μg/L 范圍內,三價砷、甲基砷和五價砷3 種砷形態均有良好的的線性關系,相關系數均優于0.999,在色譜進樣量為100 μL 的條件下,3 種砷形態的檢出限分別為 0.31 μg/L、0.52 μg/L 和 0.36 μg/L,低于文獻報道[22]的離子色譜—氫化物發生原子熒光法測定海水中砷形態的檢出限(1.00~2.34 μg/L),可滿足痕量檢測的需求。
2.6 海水樣品的測定、精密度及加標回收
對實際海水樣品(鹽度為28~31.6)進行了過濾和精密度及加標回收實驗,結果見表2,海水樣品及加標樣品的砷形態測試譜圖見圖4。

表2 海水樣品中砷形態分析測定結果
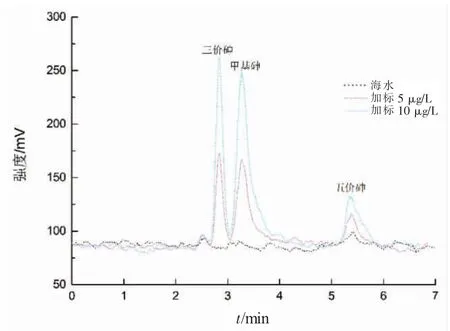
圖4 海水及加標樣品的測試色譜圖
從表2 中可以看出,海水樣品中三價砷、甲基砷的含量較低,五價砷稍高,說明海水中砷形態主要以五價砷存在,與文獻報道相一致[19-20]。對加標前后的海水樣品分別進行了測試,并進行了6 次重復測定。當加標量為5 μg/L 時,三價砷、甲基砷和五價砷的回收率在73%~96%之間,加標量為10 μg/L 時的回收率在83%~104%之間,加標回收率良好。三價砷、甲基砷和五價砷的保留時間分別為2.837 min,3.274 min 和 5.347 min,能夠在 6 min 內得到很好的分離,這與文獻報道[22]的離子色譜—氫化物發生原子熒光法在12 min 內分離測定海水砷形態相比,大大縮短了相應的分離時間,并且相對標準偏差在4.5%~8.6%之間,精密度較好。
3 結論
本文建立了液相色譜—原子熒光聯用技術測定海水中無機砷和有機砷形態的方法,在優化的實驗條件下,選用Hamilton PRP-X100 分析柱,以磷酸氫二鈉和磷酸二氫鉀為流動相(pH 為7.0),采用基體匹配法測定,三價砷、甲基砷和五價砷可在6 min 內得到有效分離。該方法檢出限低,精密度高、加標回收率良好,樣品不需要復雜的前處理,操作簡便,是測定海水及濃鹽水中砷形態的有效方法,為開展海洋環境的污染監測及海水中砷形態的遷移轉化研究提供了重要手段。
