基于1H-NMR、31P-NMR的三苯基膦三間磺酸鈉定量分析研究
2020-08-21 14:02:34梁春杰孟慶春徐曉婷柴曉飛董昭蘋呂印美王岳華蔡穎輝
分析測試學報 2020年8期
梁春杰,孟慶春,徐曉婷,柴曉飛,董昭蘋,呂印美,王岳華,蔡穎輝
(黃河三角洲京博化工研究院有限公司,山東 濱州 256500)

圖1 TPPTS的分子結構Fig.1 Molecular structure of TPPTS
三苯基膦三間磺酸鈉(TPPTS)是一種重要的化工中間體(分子結構式如圖1所示),其與金屬銠(Rh)配位后可作為羰基合成的催化劑,用于生產比原料烯烴多1個碳原子的醛。與其他催化體系相比,Rh-TPPTS具有選擇性好、催化活性高、分離回收易等優點,在正丁醛、正戊醛等醛類化合物的生產工藝中得到廣泛應用[1-2]。
TPPTS在制備過程中可能生成多種副產物,給TPPTS的分離分析帶來困難。其副產物主要包括以下3類:①苯環上磺酸基的取代位置不在間位;②三苯基膦上磺酸基的取代個數不均一,有可能生成三苯基膦二間磺酸鈉以及三苯基膦一間磺酸鈉;③TPPTS上的磷原子容易被氧化,生成三苯基氧膦三間磺酸鈉(OTPPTS)[3]。由于反應過程中副產物較多,且部分副產物不易分離,至今仍未獲得高純度的TPPTS標準品,難以通過常規液相色譜、氣相色譜等方法進行定量分析。而研究表明三苯基膦上的磺酸基位置和數目是影響催化性能的重要因素,且TPPTS被氧化為OTPPTS后其催化性能會受影響[2-4]。因此研究TPPTS合成體系中的副產物種類以及TPPTS在體系中的精確含量,對于后續Rh-TPPTS的催化性能評價與工藝優化具有重要指導意義。早期文獻通過元素分析方法來驗證TPPTS的產率[5],陳曉華等[6]則通過碘量法測定體系中的三價膦以計算TPPTS含量,但這兩種方法不能排除上述①類副產物的影響。截至目前仍未有詳細、系統的TPPTS定量分析方法的報道。
近20年來,隨著硬件設備的提升以及新型脈沖序列的不斷開發,核磁定量逐步在制藥、農化、材料科學等領域得到廣泛應用[7-12]。與傳統氣相色譜、高效液相色譜等儀器相比,核磁定量的主要優勢在于:①無需待測組分的標準品。②無需對混合體系進行分離,只要待測樣品的核磁譜圖中有1個信號峰的分離度較好,即可滿足定量分析的要求[13]。此外,隨著技術的不斷進步,核磁定量的準確度也逐步達到甚至超過了傳統色譜的定量分析水平[14-17]。
本文采用核磁共振內標法對TPPTS的定量分析進行探究。通過溶劑的選擇優化,得到了分辨率高、適于定量分析的一維核磁氫譜;并通過對TPPTS的1H、13C、31P化學位移歸屬以及縱向弛豫時間(T1)的測定,建立了TPPTS基于1H-NMR、31P-NMR的定量分析方法。兩種方法的測定結果一致性高、平行性好,且準確有效。
1 實驗部分
1.1 儀器與試劑

1.2 1H-NMR譜峰優化
稱取約10 mg TPPTS,分別用DMSO-D6、DMF-D7、D2O-D2溶解,采集1H-NMR,關鍵采樣參數設置如下:脈沖序列:zg30;采樣點數:64 K;譜寬:15 ppm;弛豫延遲時間(D1):2 s;采樣次數:16;譜中心(O1):6.5 ppm。
1.3 1H、13C、31P化學位移歸屬
以DMF-D6作為溶劑,分別采集TPPTS的一維1H-NMR、13C-NMR、31P-NMR以及二維1H-1H COSY、1H-13C HSQC、1H-13C HMBC,分別對TPPTS的1H、13C、31P化學位移進行歸屬。
1.4 縱向弛豫時間(T1)測定
T1的測定均采用t1ir脈沖序列,分別選取10個不同的弛豫恢復時間,具體設置為0.01、0.1、0.25、0.5、1、2、3、4、5、10 s。采集的數據導入Origin 9.0進行曲線擬合。
1.5 核磁內標法定量分析
1.5.231P-NMR定量分析稱取約30 mg TPPTS,約10 mg KH2PO4溶于D2O-D2中,實驗參數中D1設為35 s,O1為-3.1 ppm。

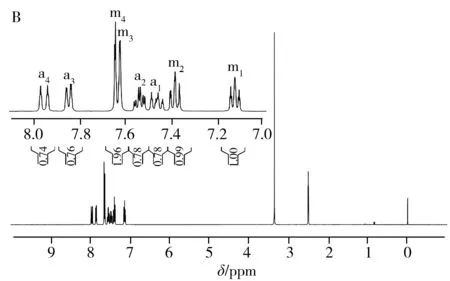
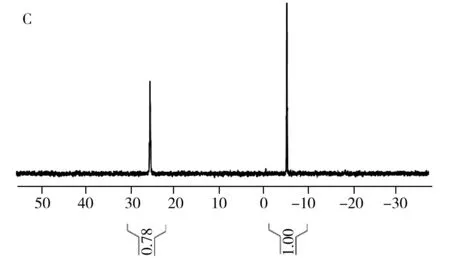
圖2 TPPTS在空氣中放置前的1H-NMR光譜(A),以及在空氣中放置24 h后的1H-NMR(B)與31P-NMR(C)光譜Fig.2 1H-NMR spectrum of TPPTS before(A) exposed to the air, 1H-NMR(B) and 31P-NMR(C) spectra of TPPTS after exposed to the air for 24 h
2 結果與討論
2.1 1H-NMR中主峰與雜質峰的區分
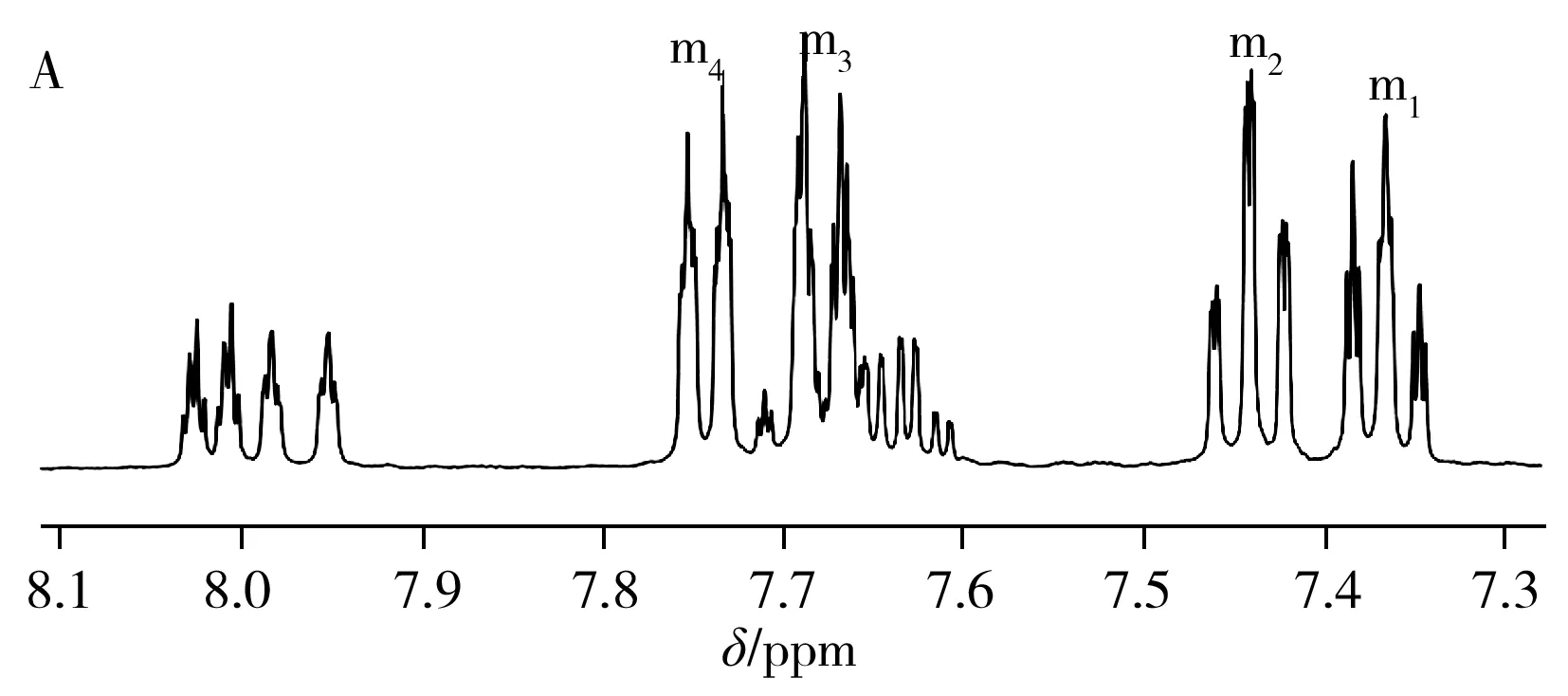
圖2A為DMSO-D6作溶劑時TPPTS的1H-NMR譜圖,圖中除溶劑峰(δ2.5 ppm)和殘余水峰(δ3.3 ppm)外,主要還包含m1、m2、m3、m4四組主峰以及a1、a2、a3、a4其他四組峰。如果把其中1個主峰(如m1)的峰面積定為1,a1~a4四組峰的峰面積均在0.4左右。上述樣品在空氣中暴露24 h后再次采集一維氫譜,結果如圖2B所示,a1~a4四組峰相對于主峰的峰面積較之前變大(由0.40變為0.78左右),說明a1~a4四組峰可能是由于TPPTS被氧化成OTPPTS,苯環上的氫化學位移向低場移動所致;對空氣中放置24 h后的樣品進行31P-NMR采集,可觀察到譜圖主要包含2個峰,且低場區較高場區的相對峰面積為0.78(見圖2C),基本與圖2B氫譜中相對峰面積一致,進一步表明譜圖中的雜質峰是由TPPTS上的三價磷原子被氧化成五價磷原子所致。
2.2 氘代試劑的優化
從圖2A可以看到,以DMSO-D6作溶劑時,TPPTS的1H-NMR在δ=7.13 ppm處的峰具有較好的分離度,可滿足核磁內標法定量的基本要求。但考慮到TPPTS在DMSO-D6中的溶解度很小,在樣品稱量過程中可能存在較大的相對誤差,為此分別采用溶解度較大的D2O-D2以及DMF-D7作溶劑采集了TPPTS的核磁氫譜(圖3)。結果顯示:以D2O-D2作溶劑時,TPPTS的4組主峰(m1、m2、m3、m4)分離度均不是很好,不適合做定量分析(圖3A);而以DMF-D7作溶劑時,TPPTS在δ=7.24、7.40、7.80 ppm處的3組峰(m1、m2、m3)均有較好的分離度,可用于定量分析(圖3B)。綜合以上分析,最終選擇DMF-D7作樣品溶劑進行核磁氫譜定量分析。相對于1H-NMR,31P-NMR的譜峰簡單且分離度好,因此,本實驗選擇溶解度較好的D2O-D2作為溶劑,采用磷譜法對TPPTS進行定量分析。
2.3 TPPTS的1H、13C、31P化學位移歸屬
為了進一步驗證上述氫譜中的四組主峰為TPPTS而非其他副產物產生,對TPPTS的1H、13C、31P進行化學位移歸屬驗證,其1H-1H COSY、1H-13C HSQC、1H-13C HMBC信號均可與TPPTS的各原子相匹配(信號歸屬見圖4),由此表明1H-NMR中的四組主峰確實由TPPTS產生,且根據主峰間的相對峰面積可判斷定量分析時所選取的信號峰是否存在其他雜質信號干擾(如圖3B中m1信號峰若有其他雜質信號重疊,那么m1與m2、m3的峰面積之比將大于1),然后通過選取合適的信號峰以保證定量分析結果的準確性。TPPTS的具體化學位移歸屬如表1所示。
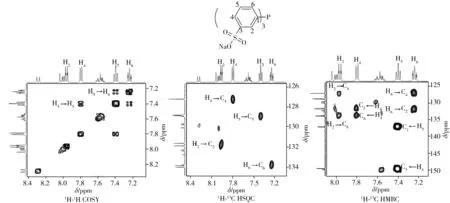
圖4 TPPTS的1H-1H COSY、1H-13C HSQC及1H-13C HMBC譜圖信號歸屬Fig.4 Signal assignments of TPPTS by 1H-1H COSY、1H-13C HSQC and 1H-13C HMBC spectra
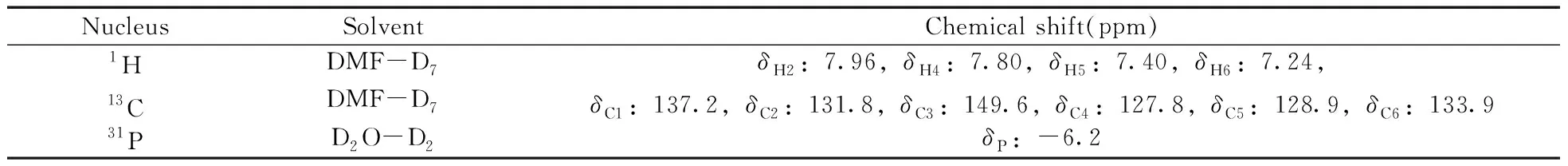
表1 TPPTS的化學位移歸屬Table 1 Chemical shift assignments of TPPTS
2.4 核磁內標法定量
2.4.1 定量分析原理核磁共振定量的基本原理是在一定條件下體系中的原子核不論所處的化學環境是否相同,單個原子核所產生的峰面積基本相等。因此,只要保證目標化合物中有1個信號峰與其他信號不重疊,再選擇1個出峰合適且已知濃度的內標化合物,即可實現目標化合物的定量分析。
假設實驗過程中內標化合物的稱取質量為m1、相對分子量為M1、質量分數為x1,積分峰面積為S1,積分區包含的原子個數為n1;假設目標化合物的稱取質量為m2,相對分子量為M2,質量分數為x2,積分峰面積為S2,積分區包含的原子個數為n2。根據上述定量原理可得:
2.4.21H-NMR定量分析核磁氫譜定量的優勢在于靈敏度高,檢測時間較短。為了盡可能達到定量原理部分所提到的單個原子核所產生的峰面積相同,需注意以下方面:①多次掃描測量時需確保弛豫延遲時間(D1)大于或等于樣品及內標化合物中所檢測原子核T1的5倍。②為消除射頻脈沖激發偏共振效應,應該把譜中心(O1)置于樣品峰及內標峰的中間。③對于有機小分子化合物的定量測試,可在溶解范圍內適當增加稱樣質量,從而減少稱量誤差。
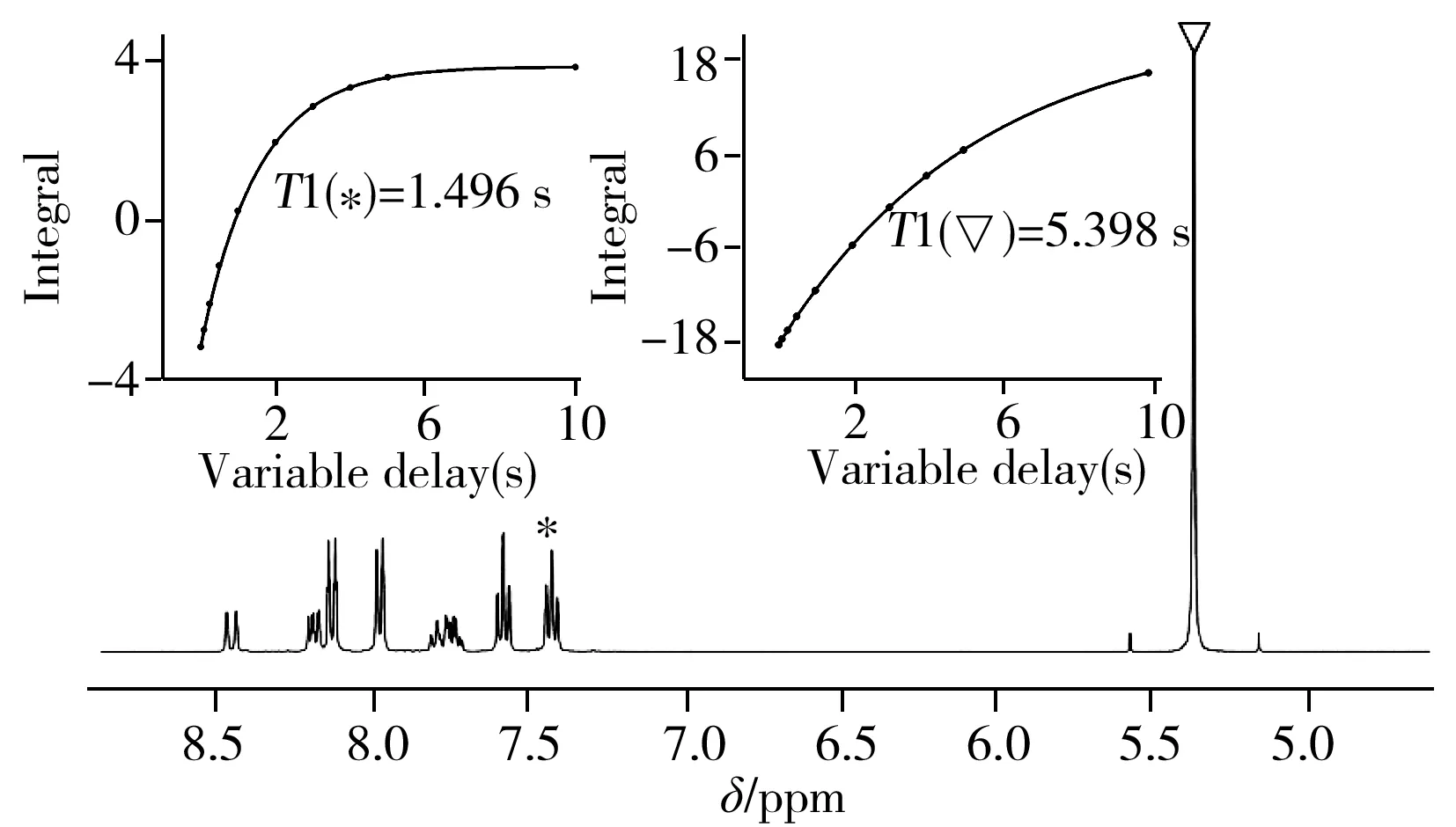
圖5 TPPTS和三烷在DMF-D7體系中的1H-NMR譜圖Fig.5 1H-NMR spectra of TPPTS and trioxane in DMF-D7insert:T1 fitting curve of the spin nuclear in TPPTS and trioxane(TPPTS和內標峰的縱向弛豫時間T1擬合曲線)
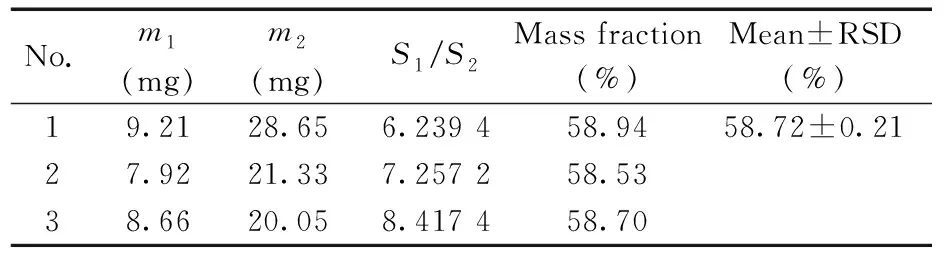
表2 TPPTS的1H-NMR定量分析結果Table 2 Quantitative analysis results of TPPTS using 1H-NMR
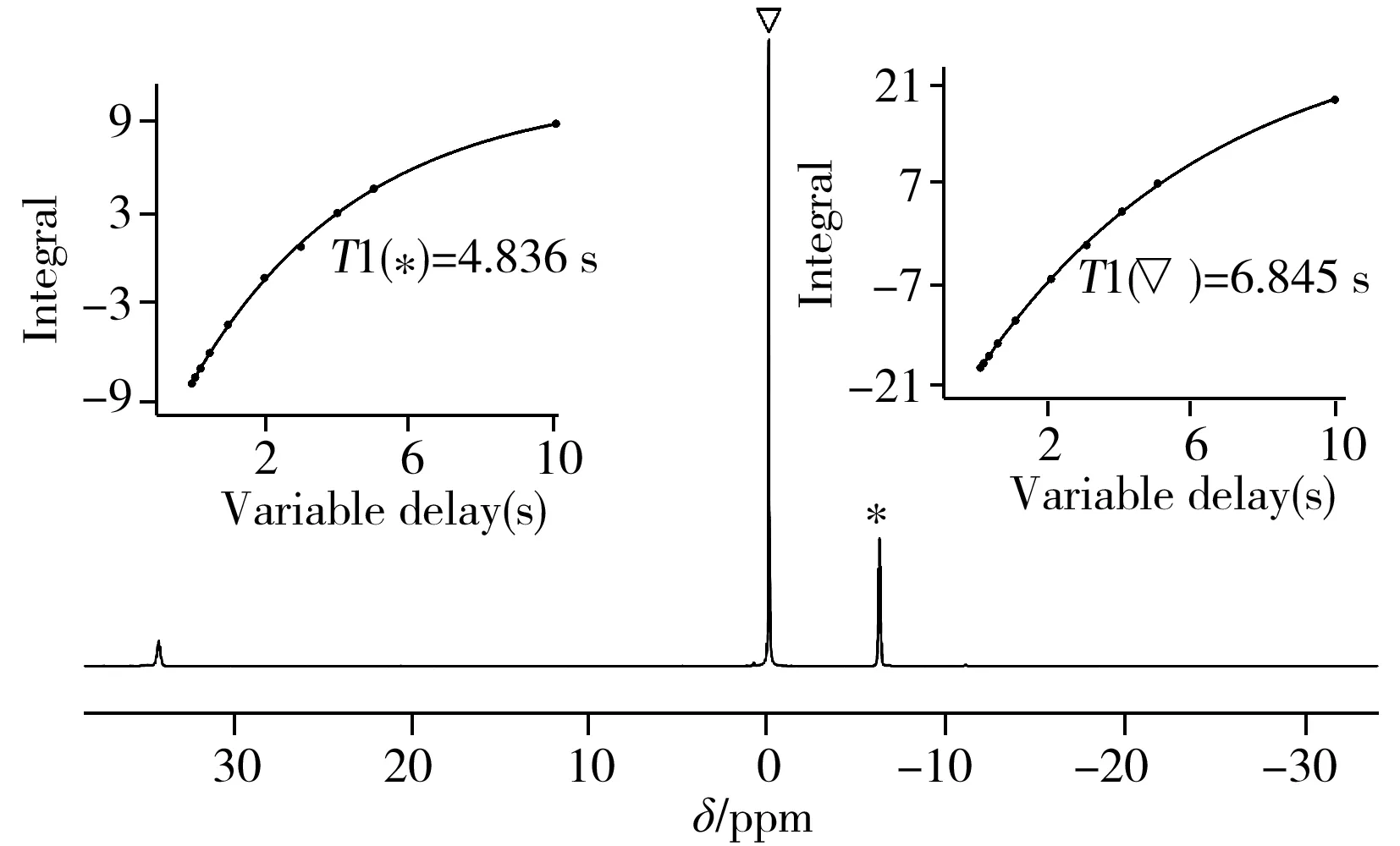
圖6 TPPTS和KH2PO4在D2O-D2體系中的31P-NMRFig.6 31P-NMR spectra of TPPTS and KH2PO4 in D2O-D2insert:T1 fitting curve of the spin nuclear in TPPTS and KH2PO4(TPPTS和內標峰的縱向弛豫時間T1擬合曲線)
2.4.331P-NMR的定量分析31P的自旋量子數為1/2,自然豐度為100%,具有較好的信號響應。雖然31P的靈敏度不如1H,但31P-NMR的化學位移分布范圍更寬,而且很多有機分子只含1種磷原子,所以譜峰重疊的概率很低,特別適合用作復雜體系中某種化合物的定量分析[18]。鑒于實際生產過程中TPPTS可能存在多種副產物,會導致氫譜譜峰嚴重重疊而無法定量,因此對TPPTS的31P-NMR定量方法進行了研究,并將其定量結果以與1H-NMR定量結果相互驗證。
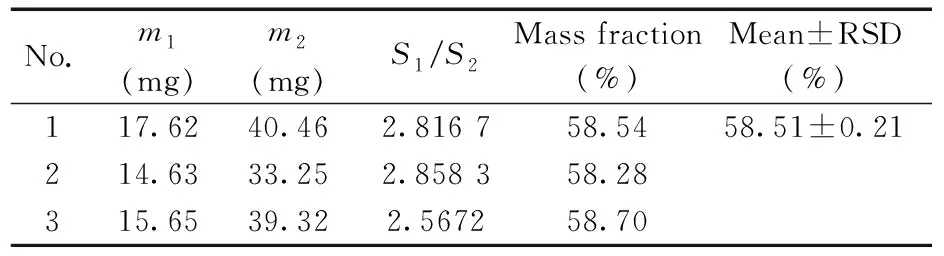
表3 TPPTS的31P-NMR定量分析結果Table 3 Quantitative analysis results of TPPTS using 31P-NMR
TPPTS的磷譜定量選取KH2PO4作為內標,圖6為TPPTS和KH2PO4在D2O-D2體系中所采集的31P-NMR以及T1擬合曲線,其中把KH2PO4的化學位移δ定為0 ppm(▽標記),此時TPPTS的化學位移在δ=-6.2 ppm(*標記);經測定得到TPPTS磷原子的T1為4.836 s,內標磷原子的T1為6.845 s。為保證自旋體系的縱向磁化強度充分恢復,D1設置為35 s(盡管分析過程中采用30°脈沖進行激發)。另外,為了盡量減少偏共振效應,設置O1=-3.1 ppm。從表3中可看出3次測量測平均值為58.51%,相對標準偏差為0.21%,最終檢測結果可表示為(58.51±0.21%)。測量結果偏差較小,且平均值與1H-NMR定量分析結果的平均值接近,僅相差0.21%,兩種方法的相互驗證說明TPPTS定量分析結果具有較高的準確性。
3 結 論
本文通過1H-NMR及31P-NMR技術建立了混合體系中TPPTS的有效定量分析。兩種方法的結果接近(平均值相差0.21%),且各有優勢:1H-NMR的定量分析耗時較短,而31P-NMR譜圖的重疊概率小,解析簡單,適合復雜混合體系中TPPTS的定量分析。本方法通過對文中定量分析過程以及一些關鍵參數進行設置,為其他化合物的定量分析提供了方法借鑒,對TPPTS的合成工藝以及Rh-TPPTS催化體系生產工藝的優化改進也具有重要指導意義。
猜你喜歡
鴨綠江(2021年35期)2021-04-19 12:24:18
新世紀智能(數學備考)(2020年11期)2021-01-04 00:38:16
考試與評價·高一版(2020年6期)2020-11-02 02:45:24
電子制作(2018年11期)2018-08-04 03:25:42
科技知識動漫(2017年5期)2017-05-11 21:34:16
科技知識動漫(2017年4期)2017-04-15 22:24:55
科技知識動漫(2017年2期)2017-02-06 20:59:46
科技知識動漫(2016年10期)2016-10-18 20:35:00
鑿巖機械氣動工具(2016年3期)2016-03-01 04:00:25
新高考·高一物理(2014年1期)2014-09-18 01:26:07
