同位素稀釋高效液相色譜-串聯質譜法測定水體、底泥、魚及蝦中氯硝柳胺殘留量
2020-08-22 04:02:02劉永濤沈丹怡楊秋紅楊移斌艾曉輝
分析測試學報 2020年8期
劉永濤,沈丹怡,董 靖,楊秋紅,楊移斌,周 順,艾曉輝*
(1.中國水產科學研究院 長江水產研究所,湖北 武漢 430223;2.農業農村部水產品質量安全控制重點實驗室,北京 100141;3.湖北省水產品質量安全工程技術研究中心,湖北 武漢 430223;4.浙江省慈溪市水產技術推廣中心,浙江 慈溪 315300)
氯硝柳胺(Niclosamide,NIC),又名殺螺胺、貝螺殺、清塘凈等,是世界衛生組織(WHO)推薦使用的滅螺劑。在我國氯硝柳胺主要用于殺滅血吸蟲傳播的中間宿主釘螺,自1992年世界銀行貸款中國血吸蟲病控制項目實施以來,每年用藥量約3 200 t以上,大量氯硝柳胺在江湖洲灘長期反復使用[1]。氯硝柳胺在我國也被批準用作清塘劑殺滅釘螺、椎實螺和野雜魚等[2]。研究表明,氯硝柳胺對魚類、蚤類、蝌蚪、螺類等具有明顯毒性[3]。Abreu等[4]通過研究氯硝柳胺與小牛胸腺DNA的相互作用,證實氯硝柳胺能引起DNA的損傷。Ostrosky-Wegman等[5]研究發現4個人類血液樣品中有2個樣品的碎屑形成與氯硝柳胺呈劑量相關增長。氯硝柳胺在人體內也能誘導產生血管舒張相關的副作用[6]。持續大范圍、大劑量使用氯硝柳胺對生態環境和消費者身體健康的影響日益受到人們關注,同時在水產養殖中氯硝柳胺的投毒案件也時有發生[7],因此,建立一種對環境水體、底泥、魚和蝦中氯硝柳胺準確定性定量的分析方法是十分必要的。
目前對環境水體中氯硝柳胺的測定方法主要有高效液相色譜法[8]、衍生化氣相色譜-質譜法[7]和液相色譜-串聯質譜法[9-11];底泥中氯硝柳胺的測定方法有加速溶劑萃取/液相色譜-串聯質譜法[12];水產品中氯硝柳胺的測定方法主要有氣相色譜法[13]、高效液相色譜法[14]和液相色譜-串聯質譜法[15-17]。氣相色譜法與氣相色譜-質譜法需進行硅烷化衍生處理,且樣品前處理過程繁瑣;液相色譜法分析氯硝柳胺易出現假陽性,且檢出限高;液相色譜-串聯質譜法靈敏度高,但存在明顯的基質效應,且不同基質的基質效應差別較大。同位素稀釋結合質譜技術是有效減少基質效應的方法[18-19]。因此,本文以氯硝柳胺的穩定同位素標記物氯硝柳胺-13C6(NIC-13C6)為內標,建立了檢測環境水體、底泥、魚和蝦中氯硝柳胺的同位素稀釋高效液相色譜-串聯質譜法。與前述文獻方法相比,該方法具有靈敏度和準確度高、樣品制備簡單、無基質效應等優點。
1 實驗部分
1.1 儀器與試劑
TSQ Quantum Access Max液相色譜-質譜聯用儀(Thermo Fisher Scientific公司);HITACHI 20PR-520型自動高速冷凍離心機(日本日立公司);Mettler-Toledo AE-240電子天平(梅特勒-托利多公司);FS-1高速勻漿機(華普達教學儀器有限公司);調速混勻器(上海康華生化儀器制造廠);氮吹儀(杭州奧盛儀器有限公司);HQ-60-Ⅱ 旋渦混合器(北京北方同正生物技術發展有限公司);HGC-12氮吹儀(上海HENGAO T&D公司)。
氯硝柳胺標準品(純度≥96%,德國Dr.Ehrenstorfer公司)、氯硝柳胺-13C6-水合物標準品(純度≥99.5%,德國Witega公司);十八烷基硅膠(C18)、乙二胺-N-丙基硅烷(PSA)(40~60 μm,天津博納艾杰爾科技有限公司);多壁碳納米管(MWCNTs)(外徑:8~15 nm,純度>95%,長度<50 μm,北京博宇高科新材料技術有限公司);乙腈、甲醇、水(色譜純,美國J.T.Baker公司),氨水、無水硫酸鈉、氯化鈉(分析純,上海國藥集團化學試劑有限公司);0.1%氨水乙腈:1 000 mL乙腈中加入2 mL 50%氨水,混勻即得。
1.2 標準溶液的配制
分別準確稱取適量氯硝柳胺、氯硝柳胺-13C6-水合物標準品,用乙腈分別溶解并定容,配制成100 μg/mL的標準儲備液,于-20 ℃冰箱中保存。分別用乙腈-水(70>∶30,體積比)將標準儲備液稀釋成1.0 μg/mL的標準中間液,于4 ℃冰箱中保存。
用乙腈-水(70>∶30)稀釋標準中間液,得到含氯硝柳胺質量濃度分別為0.2、0.5、1.0、5.0、10、20、50、100、200 μg/L,且含氯硝柳胺-13C6質量濃度均為10 μg/L的溶劑標準工作溶液。
采用“1.3”的方法制備水樣、底泥、黃顙魚自然比例帶皮肌肉和克氏原螯蝦肌肉空白樣品溶液,用空白樣品溶液稀釋標準中間液,得氯硝柳胺質量濃度分別為0.2、0.5、1.0、5.0、10、20、50、100、200 μg/L,且含氯硝柳胺-13C6質量濃度均為10 μg/L的空白基質匹配標準溶液。
1.3 樣品前處理
1.3.1 水樣移取200 mL水樣于500 mL分液漏斗中,加入10 μL 1.0 μg/mL的內標溶液,再加入8 g氯化鈉,振搖分液漏斗使其溶解。加入氨水調節水樣的pH為9.5±0.5,再加入40 mL乙酸乙酯劇烈振蕩混勻2 min,靜置30 min,取上層乙酸乙酯層于雞心瓶中,用乙酸乙酯重復提取1次。將乙酸乙酯層合并于雞心瓶中,置于40 ℃旋轉蒸發儀上旋轉蒸發至干,用4 mL乙腈分2次溶解殘渣并轉移至10 mL離心管中,置于50 ℃氮吹儀上氮吹至干,加入1 mL乙腈-水(70>∶30)定容,渦旋振蕩30 s,7 000 r/min離心5 min,過0.22 μm尼龍有機濾頭后待分析。
1.3.2 底泥去除底泥樣品中的樹枝、石頭等雜物后烘干。稱取5 g粉碎的干燥樣品于50 mL離心管中,加入3 mL蒸餾水,渦旋振蕩1 min,再加入20 μL 1.0 μg/mL的內標溶液和7 mL 0.1%氨水乙腈,渦旋振蕩30 s,再加入3 g無水硫酸鎂渦旋振蕩1 min,7 000 r/min離心5 min,轉移提取液至15 mL離心管中,重復提取1次,合并提取液,用乙腈定容至15 mL,再加入300 mg C18粉,渦旋振蕩30 s,7 000 r/min離心5 min。準確移取7.5 mL提取液至10 mL離心管中,置于50 ℃氮吹儀上氮吹至干,用1 mL乙腈-水(70>∶30)溶解殘渣,渦旋振蕩30 s,7 000 r/min離心5 min,取離心后的液體過0.22 μm尼龍有機濾頭后待分析。
1.3.3 魚、蝦樣品分別稱取5 g黃顙魚自然比例帶皮肌肉和克氏原螯蝦肌肉樣品,置于50 mL離心管中,依次加入20 μL 1.0 μg/mL的內標溶液和7 mL 0.1%氨水乙腈,后續步驟與“1.3.2”相同。
1.4 色譜與質譜分析條件
色譜條件:色譜柱:Thermo Hypersil Gold C18(150 mm×2.1 mm,5 μm);柱溫:35 ℃,流速:0.3 mL/min;流動相:A為乙腈,B為水;進樣量:10 μL。流動相洗脫梯度:0~2.0 min,50% A;2.0~2.5 min,50%~90% A;2.5~6.5 min,90%A;6.5~6.6 min,90%~50% A;6.6~7.5 min,50% A。
質譜條件:離子化模式:加熱大氣壓電噴霧離子源(HESI),選擇反應監測(SRM)模式進行檢測,負離子模式噴霧電壓:-3 000 V,源內解離電壓:0 V,蒸發氣溫度:200 ℃,鞘氣壓力:344.7 kPa,輔助氣壓力:34.5 kPa,碰撞氣及壓力:氬氣,0.195 Pa,離子傳輸毛細管溫度:350 ℃,Q1 PW 0.4,Q3 PW 0.7。
2 結果與討論
2.1 質譜條件的優化
分別將5.0 μg/mL的氯硝柳胺和氯硝柳胺-13C6標準溶液以10 μL/min的速度注射到質譜儀,分別對其母離子進行掃描,發現氯硝柳胺和氯硝柳胺-13C6在負離子模式下產生更多的[M-H]-分子離子,這與兩者含有高電負性的氯原子有關[18],母離子分別為m/z324.9和331.0,因此選擇在負離子模式下進行分析。然后以子離子掃描方式進行二級質譜分析,分別找出其定量和定性子離子及對應的碰撞能量,結果見表1。氯硝柳胺的產物離子分別為m/z289.0和171.1,分別由氯硝柳胺損失1個HCl和[324.9-HCl]-損失1個C7H3O2得到;氯硝柳胺-13C6則損失1個HCl產生m/z294.9的子離子和[331.0-HCl]-損失1個C7H3O2產生m/z176.9的子離子。
與電噴霧離子源(ESI)相比,HESI采用設定蒸發氣溫度來提高輔助氣溫度進而起到加熱的作用,因而離子化效率和重現性比ESI源好,且靈敏度有所提高[19]。本研究比較了HESI不同蒸發氣溫度(50、100、200、350、450 ℃)對10 μg/L目標物檢測靈敏度和鞘氣壓力的影響。結果顯示,隨著HESI蒸發氣溫度的提高,氯硝柳胺和氯硝柳胺-13C6的峰面積均有提高,鞘氣壓力也隨之增加。蒸發氣溫度為450 ℃時兩者的峰面積分別約為50 ℃時的2.2倍、2.5倍,鞘氣壓力約為50 ℃時的2.1倍。考慮到鞘氣壓力升高需要消耗更多的氮氣,同時兼顧檢測靈敏度,本實驗選擇蒸發氣溫度為200 ℃。

表1 目標化合物的SRM監測離子對及其質譜參數Table 1 Mass spectrometric parameters for SRM monitoring ion pairs of target compounds
2.2 色譜條件的優化
目前,液相色譜-質譜法分析氯硝柳胺的流動相有乙腈-0.1%氨水[9]、乙腈-0.1%甲酸的2 mmol/L乙酸銨溶液[17]、乙腈-水[8,11,15-16]和甲醇-水[10,12]。本實驗對上述流動相進行了篩選。結果表明,采用乙腈-0.1%氨水或乙腈-0.1%甲酸的2 mmol/L乙酸銨溶液作為流動相時,氯硝柳胺和氯硝柳胺-13C6的保留時間易漂移,且目標物的離子化也不穩定,導致連續兩次測定同一樣品時目標物的峰面積相差較大,與文獻研究結果相同[15-16]。而以乙腈-水為流動相比以甲醇-水為流動相時目標物的響應度高,因此最終選擇乙腈-水為流動相。
2.3 樣品前處理條件的優化
2.3.1 提取劑的選擇水樣中氯硝柳胺的提取劑有非離子表面活性劑Triton X-114[8]、離子對試劑[C8MIM][PF6]-乙腈[9]、乙腈-二氯甲烷[11]和三氯甲烷[13]。但Triton X-114和[C8MIM][PF6]的價格較高,且可處理的水樣體積較小導致檢出限較高;二氯甲烷和三氯甲烷的毒性較大,且濃縮過程中易爆沸。此外,由于氯硝柳胺的pKa為7.3,呈弱堿性,堿性環境有利于氯硝柳胺呈分子狀態,從而更易溶于非極性溶劑乙酸乙酯中。因此,本方法先將水樣的pH調至堿性,再以毒性小且沸點較高的乙酸乙酯作為提取劑進行提取。
廖且根等[12]采用乙腈-水作為萃取溶劑,中性氧化鋁、硅藻土作為吸附材料,利用加速溶劑萃取儀對底泥樣品中的氯硝柳胺及其降解產物進行提取。該方法需專門提取設備,提取過程較復雜。水產品中氯硝柳胺的提取劑多采用氨化乙腈[15-17],但上述文獻只給出氨化乙腈的pH值。由于乙腈為有機溶劑,用氨水調節pH時其pH值難于測定,因此比較了在乙腈中添加不同體積分數(0.05%、0.1%、0.2%、0.5%、1%)氨水對底泥、黃顙魚自然比例帶皮肌肉和克氏原螯蝦肌肉中氯硝柳胺加標(5 μg/kg)提取回收率的影響。結果表明,乙腈中添加0.1%氨水時,氯硝柳胺的提取回收率最高(約為79.7%)。因此,水樣以乙酸乙酯作為提取劑,底泥、魚、蝦以0.1%氨水乙腈作為提取劑。
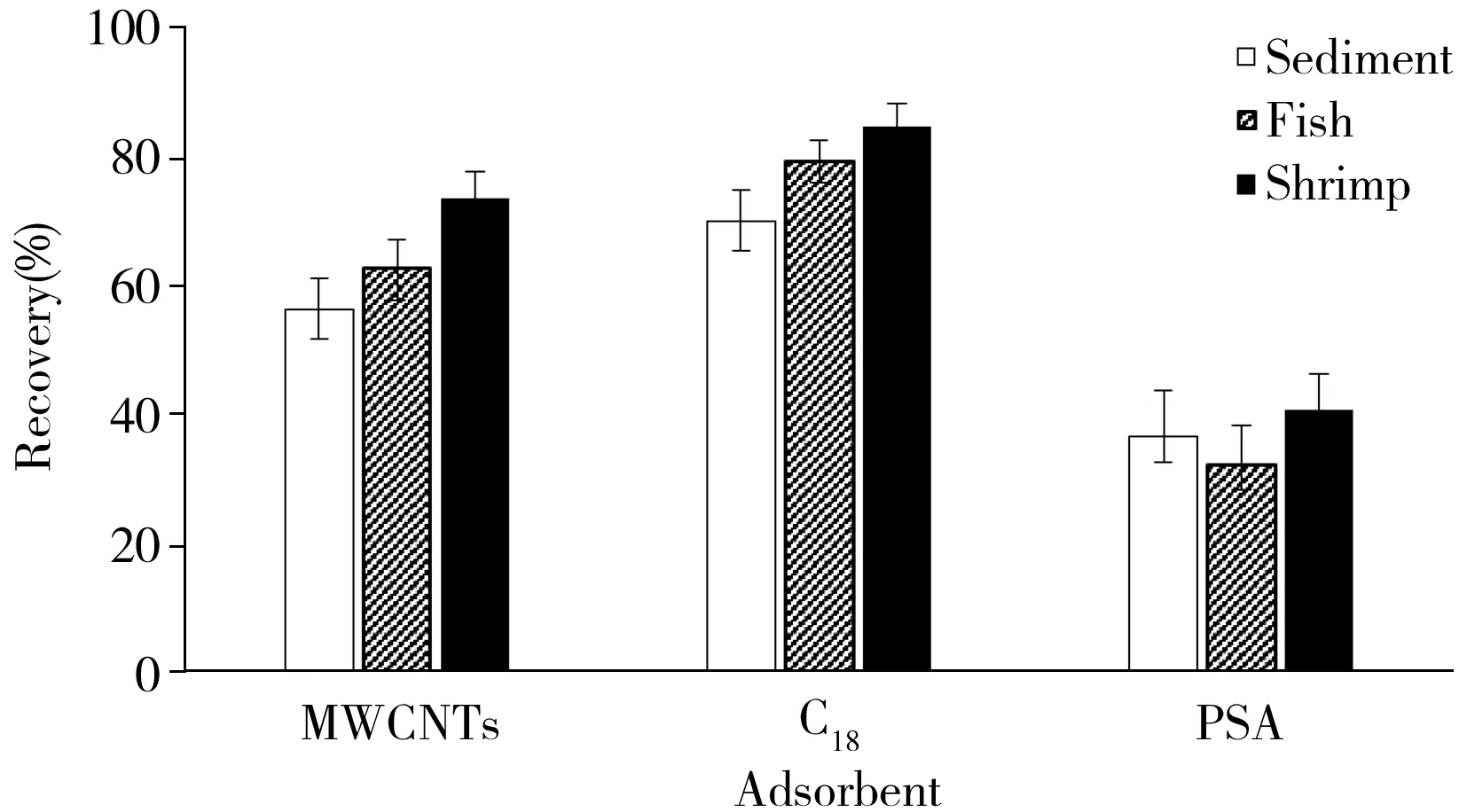
圖1 不同吸附劑對樣品提取液的凈化作用Fig.1 Effects of different adsorbents on extraction solutions
2.3.2 凈化吸附劑及其用量的選擇底泥和魚、蝦基質復雜,底泥中含有腐殖質和色素等雜質,魚、蝦含有脂肪和蛋白質等,因此樣品在提取后需進一步凈化。在底泥、黃顙魚自然比例帶皮肌肉、克氏原螯蝦肌肉空白樣品中分別添加氯硝柳胺,使其加標濃度為5 μg/kg。按照“1.3”方法進行提取后,以回收率作為評價指標,分別考察了100 mg的MWCNTs、C18和PSA對提取液的凈化效果(見圖1)。結果顯示,C18對底泥、黃顙魚、克氏原螯蝦肌肉樣品提取液的凈化效果最優,PSA最差。這是由于PSA吸附劑的硅膠上鍵合了乙二胺-N-丙基官能團,與氨基(NH2)吸附劑相似,因此對氯硝柳胺的吸附作用最大。進一步對比了不同用量C18(100、200、300 mg)對提取液的凈化效果,結果表明,C18用量為300 mg時的凈化效果最優,因此選擇300 mg C18作為凈化吸附劑。
2.4 線性關系
2.4.1 溶劑標準曲線將“1.2”制備的溶劑標準工作溶液進樣分析,以氯硝柳胺的質量濃度為橫坐標(x,μg/L),氯硝柳胺與氯硝柳胺-13C6的峰面積比為縱坐標(y),繪制溶劑標準曲線。結果表明,氯硝柳胺在0.2~200 μg/L質量濃度范圍內呈良好線性,回歸方程為y=0.192 2x+0.022 5,相關系數(r2)為0.999 5。
2.4.2 基質匹配標準曲線將“1.2”制備的水樣、底泥、黃顙魚自然比例帶皮肌肉、克氏原螯蝦肌肉空白基質匹配標準溶液進樣分析,以氯硝柳胺的質量濃度為橫坐標(x,μg/L),樣品中氯硝柳胺與氯硝柳胺-13C6的峰面積比為縱坐標(y),繪制基質匹配標準曲線。結果表明,氯硝柳胺在0.2~200 μg/L質量濃度范圍內呈良好線性,回歸方程分別為y水樣=0.188 5x水樣-0.069 6,y底泥=0.167 1x底泥-0.101 6,y魚=0.178 6x魚-0.021 7和y蝦=0.191 2x蝦+0.023 3,相關系數(r2)分別為0.999 6、0.999 8、0.999 7和0.999 9。
2.5 基質效應
液相色譜-質譜聯用技術在生物樣品分析中普遍存在基質效應。同位素內標與待測物結構相似,在色譜和質譜上的特征相似,不但可抵消質譜離子化時的基質效應,還可消除樣品前處理過程中的差異。本研究采用氯硝柳胺-13C6作為內標,以水樣、底泥、黃顙魚自然比例帶皮肌肉和克氏原螯蝦肌肉的基質匹配標準曲線與氯硝柳胺溶劑標準曲線的斜率來考察基質效應。基質效應的計算公式為[1-(基質匹配標準曲線的斜率/溶劑標準曲線的斜率)]×100%,若為負值,表明存在基質增強作用,為正值則表明存在基質抑制作用[20]。結果顯示,上述4種基質匹配標準曲線的斜率與溶劑標準曲線斜率的比值分別為0.98、0.87、0.93、0.99,表明4種基質對氯硝柳胺均為抑制作用。且斜率比值在0.8~1.2范圍內,說明4種基質對氯硝柳胺無基質效應[21]。因此,本文采用溶劑標準曲線對氯硝柳胺進行定量。
2.6 方法回收率、相對標準偏差、檢出限與定量下限
在空白水樣中添加2.5、25、250 ng/L 3個水平的氯硝柳胺,每個濃度水平做6個平行樣品,按照“1.3.1”進行樣品前處理,“1.4”條件進行測定,得3個加標水平下氯硝柳胺的平均回收率為90.5%~109%,相對標準偏差(RSD)為3.2%~11%。在空白底泥、黃顙魚自然比例帶皮肌肉和克氏原螯蝦肌肉中分別添加0.5、5.0、50 μg/kg的氯硝柳胺,每個濃度水平做6個平行樣品,按照“1.3.2”和“1.3.3”進行樣品前處理,“1.4”條件進行測定,得3種樣品的平均回收率分別為89.4%~113%、92.8%~110%和94.1%~107%,RSD分別為4.6%~12%、2.8%~11%和3.2%~9.3%。
分別以大于3倍和10倍信噪比計算檢出限(LOD)和定量下限(LOQ),氯硝柳胺在水樣中的LOD為1.0 ng/L,LOQ為2.5 ng/L,底泥、黃顙魚自然比例帶皮肌肉和克氏原螯蝦肌肉樣品的LOD為0.2 μg/kg,LOQ為0.5 μg/kg。空白黃顙魚自然比例帶皮肌肉及加標回收樣品(5 μg/kg)的SRM色譜圖見圖2。
2.7 方法應用
將該方法用于一個有大量死魚且懷疑是由氯硝柳胺中毒所致的池塘中底泥、水體和魚體樣品的測定,結果表明在送檢的池塘底泥、水體、黃顙魚和草魚自然比例帶皮肌肉樣品中檢出氯硝柳胺,其含量分別為108.5 μg/kg、12.7 μg/L、58.1 μg/kg和19.6 μg/kg。
3 結 論
本文采用同位素稀釋高效液相色譜-串聯質譜建立了環境水體、底泥、魚和蝦中氯硝柳胺的分析方法。該方法樣品前處理簡單快速,靈敏度、準確度和精密度高,可為開展氯硝柳胺在環境水體、底泥、魚、蝦中殘留量監測和富集遷移規律研究提供技術支撐,亦可為水產養殖中由氯硝柳胺引起魚體中毒事件提供一種有效的檢測識別方法。
