多壁碳納米管QuEChERS/氣相色譜-質譜聯用法快速檢測黃芪中16種農藥
2020-08-22 04:02:06崔麗麗樸向民馮志偉閆梅霞鄭培和姚麗娜
分析測試學報 2020年8期
關鍵詞:檢測
崔麗麗,樸向民,馮志偉,于 營,閆梅霞,鄭培和*,姚麗娜
(1.中國農業科學院特產研究所,吉林 長春 130112;2.撫松縣參王植保有限責任公司,吉林 長白山 134504;3.吉林省中藥材種植(養殖)重點實驗室,吉林 長春 130112)
黃芪是豆科植物蒙古黃芪Astragalusmembranaceus(Fisch.)Bge.Var.mongholicus(Bge.) Hsiao或膜莢黃芪Astragalusmembranaceus(Fisch.) Bge.的干燥根,味甘,性微溫,主要成分為皂苷、多糖和黃酮類,具有保肝利尿,補氣固表,健脾補中,抗衰老、抗疲勞,對心腦血管、保護肝臟、增強機體免疫功能等功效[1],為臨床常用藥物。建國前,黃芪主要滿足中醫臨床調劑用,隨著中成藥工業的快速發展,黃芪成為了中成藥和保健品原料,國內外市場對其需求日趨增大,我國成為黃芪的主要產地及出口國。然而,近年來中藥材農殘問題已引起國內外的廣泛關注[2],韓國、日本等對中藥材中農藥殘留限量的相關規定作了很大調整,不僅增加了農藥檢測種類,還降低了某些常用農藥殘留限量值;日本肯定列表對未制定限量標準的農藥均采取一律標準0.01 mg/kg[3],這對我國的中藥材種植中農藥的合理使用及農殘檢測技術提出了新的挑戰。目前,我國2015版《中國藥典》規定了黃芪中總六六六(α-BHC、β-BHC、δ-BHC、γ-BHC之和)和總滴滴涕(p,p′-DDE、p,p′-DDD、o,p′-DDT、p,p′-DDT之和)均不得超過0.2 mg/kg,五氯硝基苯不得超過0.1 mg/kg[4]。此外,黃芪病害有白粉病、麻口病、銹病、立枯病、紫紋羽病和根腐病等[5-7],化學防治常施用多菌靈、甲霜靈、嘧菌酯、咯菌腈等農藥[8]。因此,為了打破出口貿易壁壘,亟待建立黃芪中更多品種農藥殘留的快速檢測方法。
目前,農藥殘留檢測的提取方法有超聲法、振蕩法、均質法、固相萃取、索氏提取及超臨界流體萃取等[9-11],凈化方法有濃硫酸磺化法、液液萃取(LLE)、固相萃取凈化(SPE)、凝膠滲透色譜凈化(GPC)及QuEChERS方法等[12-15],分析技術為氣相色譜法(GC)、高效液相色譜法(HPLC)及其串聯質譜法等[16-20]。其中QuEChERS法(Quick,Easy,Cheap,Effective,Rugged,Safe)是基于分散固相萃取發展起來的集萃取和凈化為一體的新型前處理方法,已成為美國分析化學家協會標準方法(AOAC 2007.01)[21]和歐洲標準化委員會標準方法(EN 15662)[22],與SPE和LLE等方法相比能夠有效縮短前處理時間,提高檢測效率,顯著降低了有機溶劑用量,已被廣泛用于食品、蔬菜等的農殘檢測[23-27],以及食品中藥物[28-29]、真菌毒素[30]的檢測。多壁碳納米管是由多層石墨片卷曲而成的碳納米管,層與層之間呈無序排列,具有比表面積大、傳導率高、化學穩定性好、機械強度高等優點,已在農藥殘留分析中應用較多,且備受重視[31-32]。本研究根據黃芪病害防治及農藥使用等實際情況,將多壁碳納米管與QuEChERS方法相結合,建立了氣相色譜-質譜聯用測定黃芪中α-六六六、β+γ-六六六、δ-六六六、五氯硝基苯、p,p′-滴滴伊、o,p′-滴滴涕、p,p′-滴滴滴、p,p′-滴滴涕、甲霜靈、二嗪磷、七氯、嘧菌環胺、腐霉利、順式氯丹、反式氯丹、咯菌腈16種農藥殘留的分析方法,并成功用于實際黃芪樣品中16種農藥殘留的檢測。
1 實驗部分
1.1 儀器與試劑
7890A-5975C型氣相色譜-質譜聯用儀(美國安捷倫科技有限公司),ML503、ME204天平(感量分別為0.000 1 g和0.000 01 g,梅特勒公司),TGL-16M型高速離心機(湖南湘儀離心機有限公司),IKA T25均質器(德國IKA公司),LSE6776渦旋混合器(康寧公司)。
乙腈、丙酮、正己烷(色譜純,美國Fisher公司);0.22 μm有機相微孔濾膜(北京迪馬科技有限公司);氯化鈉、氯化鋰、無水硫酸鈉(分析純,北京化學廠);多壁碳納米管(MWNTs)A、B、C內徑均為5~10 nm,長度為10~30 μm,外徑從小到大分別為10~20、20~30、30~50 nm(上海麥克林生化有限公司);N-丙基乙二胺(PSA)和MgSO4(分析純,北京迪科馬科技有限公司);實驗用水為GB/T 6682規定的一級水。α-六六六、β-六六六、γ-六六六、δ-六六六、p,p′-滴滴滴、o,p′-滴滴涕、p,p′-滴滴涕、p,p′-滴滴伊、嘧菌環胺、腐霉利、五氯硝基苯、七氯、二嗪磷、順氏氯丹、反式氯丹、甲霜靈和咯菌腈農藥標準物質純度均大于99.5%,購于國家標準物質中心。黃芪于2018年9月采集于內蒙古地區,經專家鑒定為膜莢黃芪AstragalusMembranaceus(Fisch.) Bunge。
1.2 標準溶液配制
精密稱取50 mg(精確至0.01 mg)各農藥標準物質,用乙腈溶解并定容配成質量濃度均為500 μg/mL的單標儲備液,于-20 ℃冷凍儲存。精密吸取適量各農藥單標儲備液于25 mL容量瓶中用乙腈定容,使用時逐級稀釋成系列混合標準工作溶液。
1.3 樣品前處理
鮮黃芪經40 ℃后干燥后取不少于200 g試樣,置于高速粉碎機中粉碎,過100目篩3~5次后充分混勻,裝入聚乙烯塑料袋中保存。
精確稱取已粉碎均勻試樣1.0 g(精確至1 mg),加入10 mL水浸泡30 min,再加入20 mL乙腈,高速(3 000 r/min)渦旋振蕩提取3 min,加入1.5 g NaCl+6 g MgSO4混勻1 min,以5 000 r/min離心5 min,取上清液5 mL,加入到由75 mg PSA、450 mg MgSO4與20 mg多壁碳納米管A組成的混合凈化劑中,渦旋1 min,混勻,以5 000 r/min離心5 min,取上清液上機檢測。
1.4 色譜-質譜條件
色譜條件:色譜柱為HP-5ms石英毛細管柱,30 m×0.25 mm(內徑)×0.25 μm(膜厚);程序升溫:初始柱溫100 ℃,保持1 min,以30 ℃/min升至130 ℃,再以5 ℃/min升至250 ℃,保持10 min,最后以10 ℃/min升至280 ℃,保持10 min。進樣口溫度250 ℃,載氣為高純氦氣(純度>99.999%),流速1.0 mL/min;尾吹氣流量29 mL/min,進樣體積為1 μL,進樣方式為脈沖不分流進樣,脈沖壓力172 250 Pa,延時0.75 min,0.75 min后開閥。
質譜條件:離子源為電子轟擊源(EI),電離能量70 eV,離子源溫度230 ℃,四極桿溫度150 ℃,溶劑延遲時間10 min,選擇離子監測(SIM)模式,傳輸線溫度280 ℃。16種農藥的保留時間及檢測離子見表1。
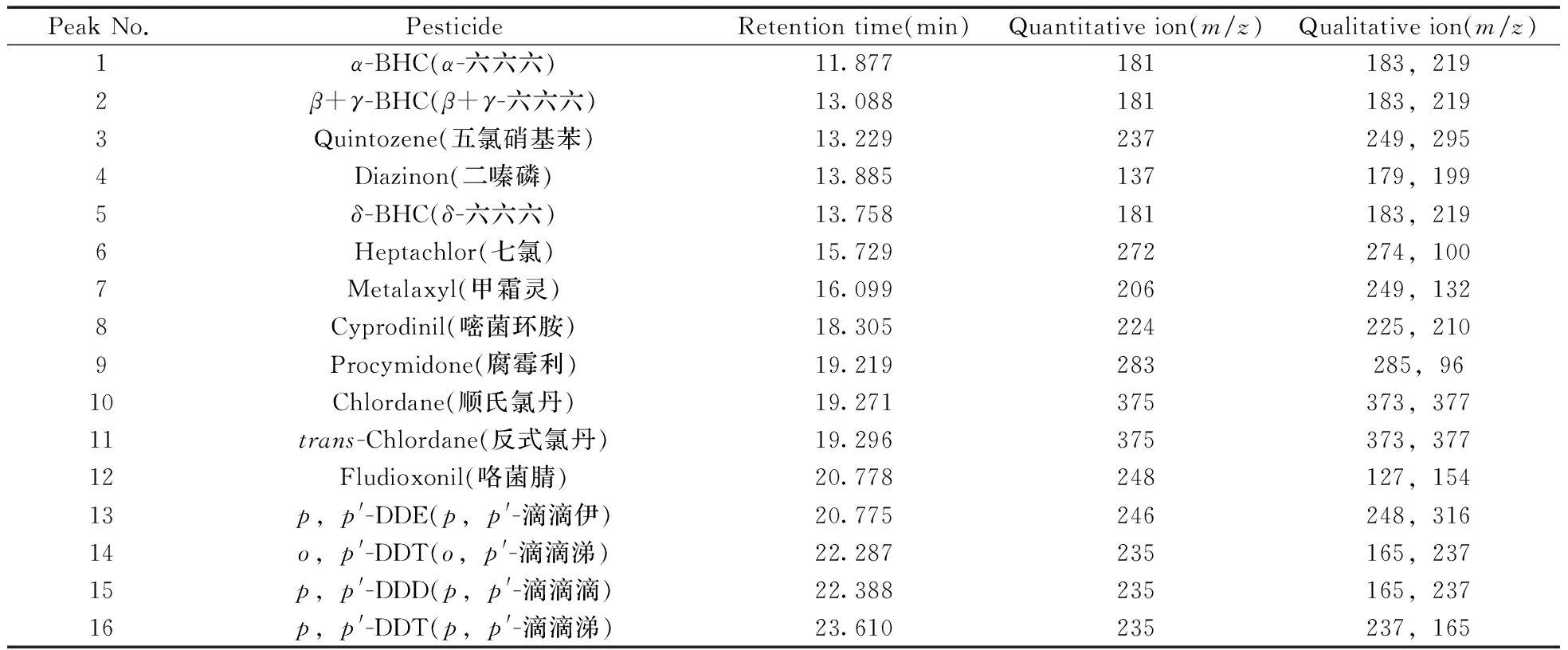
表1 16種農藥的質量分析參數Table 1 Mass spectrometric parameters for 16 pesticides
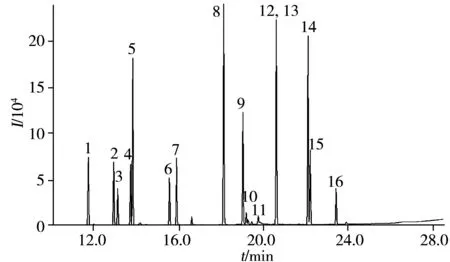
圖1 優化色譜條件下的農藥混合標準溶液的總離子流圖Fig.1 Total ion chromatogram of pesticides standard solution under optimization chromatographic conditionsthe number 1-20 were the same as those in Table 1
2 結果與討論
2.1 色譜-質譜條件優化
選擇對弱極性化合物分離較好的毛細管柱色譜柱HP-5ms,通過SCAN模式掃描,選擇掃描質量范圍為30~500 amu,確定每種農藥的保留時間及定性定量離子,并建立SIM離子監測方法。每種農藥選擇一個定量離子,2個定性離子,按照保留時間順序分段檢測,因β-六六六和γ-六六六兩個譜峰重疊,故合并計算為一種農藥,優化條件后16種的農藥保留時間、定量離子、定性離子見表1,總離子流圖見圖1。
2.2 萃取劑與萃取鹽的選擇
2.3 多壁碳納米管的選擇
向空白樣品中加入一定量農藥混合標準溶液配成0.5 mg/kg的樣品溶液,以16種農藥的回收率考察“1.1”中A、B、C 3種型號MWCNTs的凈化效果。結果顯示,A、B、C 3種MWCNTs凈化后回收率在70%~120%范圍內的農藥數量分別為13、10、7種,這是因為MWCNTs的外徑越小,其比表面積越大,吸附能力越強,因此選擇MWCNTs A進行凈化。此外,實驗還考察了MWCNTs A的用量(5、10、20、25、30、35、40 mg)對凈化效果的影響,發現其用量為20 mg時,回收率在70%~120%范圍內的農藥數量最多(13種),因此最終選擇20 mg MWCNTs A進行凈化。
2.4 凈化劑種類及用量的優化
根據本研究所涉及的農藥種類和特性,選擇N-丙基乙二胺(PSA)+無水MgSO4+ MWCNTs進行組合凈化[34],實驗考察了50 mg PSA+300 mg MgSO4+20 mg MWCNTs A、75 mg PSA+450 mg MgSO4+20 mg MWCNTs A和150 mg PSA+900 mg MgSO4+20 mg MWCNTs A的凈化效果。結果發現,75 mg PSA+450 mg MgSO4+20 mg MWCNTs A的凈化效果最好,此時農藥的回收率在70%~120%范圍內的數量最多。
2.5 基質效應
基質效應(ME=B/A,A為純溶劑中農藥的響應值,B為樣品基質中添加的相同含量農藥響應值)大于1.2為基質效應增強,小于0.9為基質效應減弱。本研究中大多數農藥均有較強的基質效應,因此選擇脈沖不分流進樣方式并結合基質匹配標準法來消除基質效應的影響[30]。另外,研究還發現,提取液體積過大時,大多數農藥的ME較大,但提取液體積過小時,農藥提取不完全,回收率較低。綜合考慮,選擇提取液體積為5.0 mL,此時所有農藥的ME和回收率均在合理范圍內。
2.6 方法學考察
分別配制質量濃度為0.01、0.02、0.05、0.1、0.5、1.0、2.0、5.0 mg/L的16種農藥混合標準工作液,在優化條件下測定。以各農藥的定量離子峰面積(y)為縱坐標,對應以質量濃度(x,mg/L)為橫坐標繪制標準曲線,分別以3倍信噪比(S/N=3)和S/N=10計算檢出限(LOD)和定量下限(LOQ)。另向空白黃芪樣品中準確加入一定量16種農藥混合標準溶液,使其添加質量分數分別為0.05、0.2、0.5 mg/kg,每個添加水平設定3個平行和1個空白對照,采用本方法測定并計算各農藥的加標平均回收率和相對標準偏差(RSD,n=3)。結果顯示,α-六六六、β+γ-六六六、δ-六六六、五氯硝基苯、p,p′-滴滴伊、o,p′-滴滴涕、p,p′-滴滴滴、p,p′-滴滴涕、甲霜靈、二嗪磷、七氯、嘧菌環胺、腐霉利、順式氯丹、反式氯丹、咯菌腈16種農藥在0.01~5.0 mg/L范圍內線性良好,相關系數為0.974~1.000,LOD為0.001~0.011 mg/kg,LOQ為0.002~0.035 mg/kg,3個加標水平下的回收率為72.1%~115%,RSD為1.2%~14%。
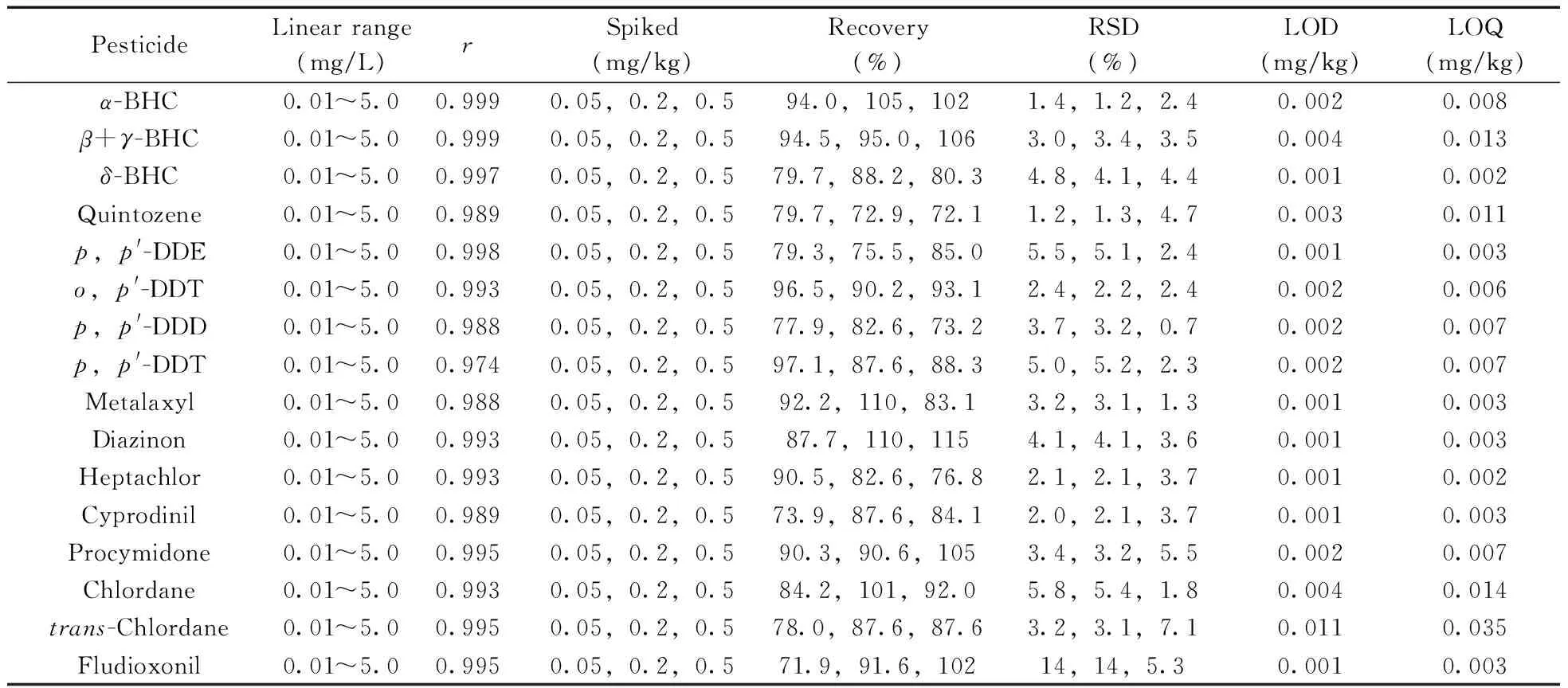
表2 黃芪中16種農藥的線性范圍、相關系數、回收率、相對標準偏差、檢出限及定量下限Table 2 Linear ranges,correlation coefficients,recoveries,RSDs,LODs and LOQs for 16 pesticides in Astragalus membranaceus
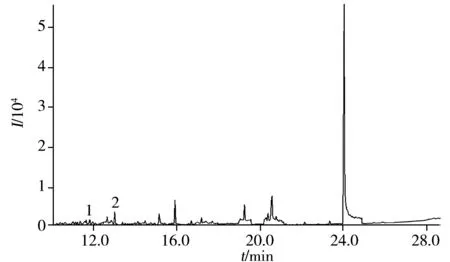
圖2 實際樣品的色譜圖Fig.2 Chromatogram of a actual sample1:α-BHC;2:β+γ-BHC
2.7 實際樣品檢測
采集內蒙古黃芪種植區內12份生長期鮮黃芪,將其烘干粉碎后在優化條件下測定。結果檢出α-六六六、β+γ-六六六,平均含量分別為0.011、0.013 mg/kg,其余農藥未檢出,其中一份黃芪的色譜圖見圖2。
3 結 論
本研究采用改進的QuChERS方法結合多壁碳納米管凈化,建立了中藥材黃芪中六六六、滴滴涕、五氯硝基苯等16種農藥的GC-MS分析方法。該方法的平均回收率為71.9%~115%,RSD為1.2%~14%,完全符合農藥殘留分析的要求,具有快速、準確、環保的優點,極大地提高了檢測效率,也減少了對環境的污染,保障了檢驗人員的健康。
猜你喜歡
中國設備工程(2022年12期)2022-07-11 04:33:00
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2019年9期)2019-11-25 07:34:36
中學生數理化·七年級數學人教版(2019年9期)2019-11-25 07:34:34
中學生數理化·七年級數學人教版(2019年12期)2019-05-21 02:53:50
中學生數理化·七年級數學人教版(2019年12期)2019-05-21 02:53:48
