Btk抑制劑依魯替尼對高糖刺激巨噬細胞炎癥的抑制作用及機制
2020-09-09 08:21:14許思敏吳永貴
安徽醫科大學學報 2020年8期
關鍵詞:檢測
許思敏,范 哲,吳永貴
糖尿病腎病(diabetic nephropathy, DN)是糖尿病重要的微血管并發癥之一。研究[1]表明DN的發生發展與炎癥有很大關系,其中腎臟組織巨噬細胞的浸潤與腎臟組織炎癥有著密切的聯系。Bruton酪氨酸激酶(Bruton's tyrosine kinase, Btk)是一種非受體型酪氨酸激酶,研究[2]表明Btk不僅參與了獲得性免疫調節,在固有免疫調節中也起到了重要作用,例如產生炎癥因子單核細胞趨化蛋白1 (monocyte chemo-attractant protein-1, MCP-1),腫瘤壞死因子α (tumor necrosis factor-alpha, TNF-α),白細胞介素1β (interleukin-1beta, IL-1β)[3]。此外,研究[4]表明Btk活性可以通過與Toll樣受體(如TLR2和TLR4),相互作用后產生,增強NF-κB p65亞基的激活作用。總之,Btk在炎癥和免疫反應中發揮重要作用。
依魯替尼(ibrutinib),又名PCI-32765,是一種高度選擇性的小分子Btk抑制劑,通過共價結合半胱氨酸-481 (cysteine 481, Cys-481)抑制Btk活性[5]。PCI-32765近年來在臨床前研究和臨床試驗中均顯示出很強的抗淋巴瘤活性[6],此外,在動物自身免疫性疾病模型,如類風濕性關節炎中也有使用[7]。該研究主要探討PCI-32765對高糖刺激巨噬細胞釋放炎癥因子的影響及其機制。
1 材料與方法
1.1 抗體與試劑葡萄糖、甘露醇購自美國Sigma公司;Btk抑制劑PCI-32765購自美國Selleck公司;試劑盒試劑購自美國Invitrogen公司;SYBR Green熒光定量PCR Master Mix試劑盒、Revert Aid第一鏈cDNA合成試劑盒、CCK-8試劑盒均購自南京諾唯贊生物科技有限公司;FITC標記的抗小鼠F4/80抗體、APC標記的抗小鼠/抗人CD11b抗體及同型對照均購自美國Bio Legend公司;Btk抗體、細胞外信號調節蛋白激酶(extracellular regulated protein kinases, ERK)抗體、c-Jun氨基末端激酶(c-Jun N-terminal kinase, JNK)抗體、p38絲裂原活化蛋白激酶(mitogen-activated protein kinase, MAPK)抗體、核因子κB(nuclear factor kappa B, NF-κB) p65抗體和核轉錄因子抑制蛋白(inhibitor of kappa B, IκB)抗體及其相應磷酸化產物p-Btk抗體、p-ERK抗體、p-JNK抗體、p-p38MAPK抗體、NF-κB p-p65抗體和p-IκB抗體以及誘導型一氧化氮合酶(inducible nitric oxide synthase, iNOS)抗體均購自美國Cell Signaling Technology公司;β-actin抗體、辣根過氧化物酶標記的抗兔IgG抗體和抗鼠IgG抗體購自武漢三鷹生物技術有限公司;蛋白質檢測試劑盒購自江蘇碧云天生物技術研究所;檢測鼠IL-1β的ELISA試劑盒均購自廣州瑞博奧生物科技有限公司;檢測鼠MCP-1、TNF-α的ELISA試劑盒購自上海依科賽生物科技有限公司;ECL發光液購自美國Thermo Scientific公司。
1.2 骨髓來源巨噬細胞(bonemarrow-derivedmacrophages,BMMs)的培養BMMs由雄性C57小鼠分離獲得。選用6~8周齡雄性C57小鼠的股骨和脛骨,去除肌肉及結締組織,用75%乙醇浸泡消毒3 min后,將股骨和脛骨移至4 ℃預冷的無菌PBS溶液中漂洗1 min。在超凈臺剪斷骨的兩端,用1 ml注射器緩緩將骨髓腔內細胞沖洗進含2%的胎牛血清(fetal bovine serum, FBS) 4 ℃預冷的無菌PBS溶液(約6 ml/只)中。將含有骨髓腔內細胞的沖洗液經2 290 r/min 4 ℃離心10 min,棄去上清液,加入3 ml紅細胞裂解液去除紅細胞,2 290 r/min 4 ℃ 離心5 min。將沉淀的細胞重懸于含10 %FBS,1%青鏈霉素及15% L929細胞上清液的DMEM低糖培養基(5 mmol/L)中,在37 ℃,5% CO2的培養箱中培養3 d后更換培養基,此后每天更換。7 d后采用流式細胞術檢測BMMs的成熟度及純度。
1.3 細胞活力檢測采用CCK-8試劑盒檢測細胞活力。在96孔板每個孔中加入1×104個BMMs。24 h后,使用不同濃度的PCI-32765或二甲基亞砜處理BMMs 45 min,再予以高糖刺激24 h。向每個孔中加入10 μl CCK-8溶液,繼續培養4 h。用酶標儀檢測490 nm下各孔的吸光度(OD)值。細胞活力的計算:細胞活力=(加藥細胞OD-空白OD)/(對照細胞OD-空白OD)×100%。將低糖組的平均細胞活力定為100%,其余實驗結果以百分比表示。
1.4 實驗條件的優化與實驗分組采用20~35 mmol/L高糖刺激BMMs,使用甘露醇作為滲透壓對照,選定影響Btk表達及BMMs活化的最佳濃度作為實驗中的高糖濃度。采用10-9~10-5mmol/L PCI-32765影響高糖刺激BMMs,選定抑制Btk表達的最佳濃度作為實驗中的抑制劑濃度。根據研究目的和實驗條件優化結果,將BMMs分為低糖組(LG),抑制劑對照組(LG+PCI-32765),高糖組(HG)和抑制劑組(HG+PCI-32765),其中,LG組葡萄糖濃度為5 mmol/L。
1.5 RNA提取及qRT-PCRRNA提取和qRT-PCR的操作方法參考文獻報道[8]。使用TRIzol試劑從實驗所得BMMs中提取總RNA。通過逆轉錄反應體系將1 μg RNA逆轉錄為cDNA。采用SYBR Green熒光定量PCR Master Mix試劑盒完成實時定量PCR,實現cDNA擴增。檢測mRNA的引物序列為包括:GAPDH作為內參:F: 5′-ACCCCAGCAAG GAGACACTGAGCAAG-3′, R: 5′-GGCCCCTCCTGTTA TTATGGGGGT-3′; TNF-α: F: 5′-CCCTCCTGGCCAA CGGCATG-3′, R: 5′-TCGGGGCAGCCTTGTCCCTT-3′; MCP-1: F: 5′-TTGACCCGTAAATCTGAAGCTAA T-3′, R: 5′-TCACAGTCCGAGTCACACTAGTTAC-3′; IL-1β: F:5′-GCCTCGTGCTGTCGGACCCATAT-3′, R: 5′-TC CTTTGAGGCCCAAGGCCACA-3′。使用2-ΔΔCt法計算mRNA相對GAPDH的表達量。
1.6 ELISA將BMMs根據實驗要求按組別處理后從培養基中提取上清液,5 000 r/min 4 ℃離心10 min,離心后再次留取上清液。根據ELISA試劑盒說明書檢測MCP-1、IL-1β、TNF-α。
1.7 激光共聚焦檢測將細胞鋪板于24孔板。在含有PCI-32765或不含PCI-32765的培養基中預處理45 min后,再加入高糖刺激24 h,室溫下用4%多聚甲醛固定30 min。使用混合溶液(5%驢血清+0.2%聚乙二醇辛基苯基醚)封閉2 h后,加入iNOS或NF-κB p65一抗4 ℃孵育過夜。PBS洗滌3次后,加入F4/80標記的一抗Alexa Fluor 647和二抗Alexa Fluor 488,37 ℃避光孵育2 h。再次用PBS洗滌3次后,滴入DAPI染色細胞核10 min。使用Leica TCS SP5激光共聚焦顯微鏡(Leica公司,德國)觀察巨噬細胞。
1.8 流式細胞術分析將成熟的BMMs按組別處理后分別收集細胞,每組1×106個,2 500 r/min離心5 min,棄去上清液,用PBS (500 μl/管)重懸細胞,加入CD16/CD32抗體,室溫處理20 min后,2 500 r/min離心10 min,棄去上清液,再次以PBS(500 μl/管)重懸細胞,加入FITC標記的F4/80、APC標記的CD11b,于室溫下避光孵育30 min。PBS洗滌兩次后,加入500 μl PBS,用流式細胞術檢測巨噬細胞。
1.9 Western blotBMMs在含有PCI-32765或不含PCI-32765的培養基中預處理45 min,再按實驗設計予以高糖刺激。提取各組總蛋白并轉移至PVDF膜上,以5%脫脂牛奶37 ℃封閉2 h后,加入一抗4 ℃孵育過夜。PBST洗滌3次后孵育相應二抗,孵育條件為37 ℃ 45 min。再次用PBST洗滌3次后,將PVDF膜置于ECL發光液中15~30 s后,采用化學發光凝膠成像系統檢測蛋白表達情況。最后,使用ImageJ軟件統計各條帶灰度值,并與內參比較分析。
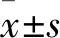
2 結果
2.1 BMMs的鑒定巨噬細胞采用FITC標記的F4/80抗體和APC標記的CD11b兩種抗體標記。如圖1A所示,F4/80及CD11b均陽性表達的細胞判定為成熟的BMMs。第7天獲得純度為94.5%的巨噬細胞。
2.2 實驗條件的優化如圖1B所示,Western blot分析結果顯示在不同濃度的高糖組中,30 mmol/L的高糖組中p-Btk和iNOS表達較5 mmol/L低糖組增加最多,差異有統計學意義(F=108.938,P<0.05;F=79.779,P<0.05),因此采用30 mmol/L作為實驗中高糖濃度。如圖1C所示,PCI-32765在10-6mmol/L和10-5mmol/L對p-Btk表達抑制效果最佳(F=4 285.000,P<0.01;F=426.276,P<0.01),且兩者之間的差異無統計學意義,故選擇較低濃度10-6mmol/L作為實驗中抑制劑對照組和抑制劑組中PCI-32765濃度。
2.3 PCI-32765對BMMs活力的影響為了確定PCI-32765是否影響BMMs活力,采用不同濃度的PCI-32765預處理巨噬細胞45 min,再加入高糖刺激24 h。如圖1D所示,與未處理細胞相比,10-8~10-5mmol/L PCI-32765無明顯細胞毒性,當PCI-32765濃度達到10-4mmol/L時,高糖刺激下的骨髓巨噬細胞活力受到影響,差異有統計學意義(F=34.287,P<0.05)。
2.4 PCI-32765對BMMs趨化的影響為了檢測PCI-32765對BMMs趨化功能的影響,將高糖誘導的BMMs分為2組,一組使用PCI-32765處理,另一組不使用PCI-32765,分別檢測細胞趨化情況。結果顯示,高糖刺激的BMMs趨化量高于低糖組(F=191.919,P<0.05),而10-6mmol/L PCI-32765預處理后,BMMs趨化受到抑制(F=18.901,P<0.05) (圖2)。

圖1 BMMs的鑒定
2.5 PCI-32765對BMMs iNOS表達的影響為了確定PCI-32765是否能改變BMMs極化,采用共聚焦顯微鏡分析BMMs的表型。結果顯示,未處理的巨噬細胞中綠色(iNOS)熒光較弱,而在高糖誘導的BMMs中,觀察到更強的iNOS染色,經PCI-32765處理后iNOS染色減弱(圖3A)。Western blot分析結果提示,高糖增加了iNOS的表達(F=173.206,P<0.01),PCI-32765降低了iNOS的表達(F=22.289,P<0.05)(圖3B)。
2.6 PCI-32765對高糖刺激的BMMs炎癥因子分泌和mRNA水平的影響用ELISA法分別檢測各組培養基上清液中TNF-α、 IL-1β、 MCP-1含量。如圖4A所示,高糖組中TNF-α、 IL-1β和MCP-1的水平高于低糖組,差異具有統計學意義(F=10.882,P<0.05;F=70.218,P<0.05;F=349.905,P<0.05)。而相比高糖組,PCI-32765抑制了高糖刺激產生的這些細胞炎癥因子,差異有統計學意義(F=30.477,P<0.05;F=22.599,P<0.05;F=370.476,P<0.05)。用qRT-PCR法分別檢測各組BMMs中TNF-α、IL-1β和MCP-1的mRNA相對表達量。如圖4B所示,高糖組中,TNF-α、 IL-1β和MCP-1的mRNA水平高于低糖組,差異有統計學意義(F=80.279,P<0.05;F=15.987,P<0.05;F=505.917,P<0.05),而PCI-32765抑制了其mRNA的表達(F=285.553,P<0.05;F=321.007,P<0.05;F=1 588.000,P<0.05)。

圖2 PCI-32765對高糖刺激BMMs趨化作用的影響

圖3 PCI-32765對高糖刺激BMMs活化的影響

圖4 PCI-32765對高糖刺激BMMs分泌促炎因子及mRNA表達的影響
2.7 PCI-32765對高糖刺激的BMMs中p-Btk、p-p38MAPK、p-ERK、p-JNK、NF-κB p-p65和p-IκB表達的影響檢測高糖刺激下,不同時間MAPK及NF-κB信號通路各蛋白表達情況,根據最佳表達時間為選取高糖刺激時長。如圖5、6所示,Western blot結果分析表明,高糖誘導的BMMs組中p-Btk表達增加(F=313.820,P<0.01),同時,p-p38MAPK、p-ERK、p-JNK和NF-κB p-p65水平亦有增加(F=3 824.000,P<0.05;F=1 083.000,P<0.05;F=6 147.000,P<0.05;F=2 833.000,P<0.05)。抑制劑對照組中上述蛋白表達相比低糖組無明顯差別,PCI-32765抑制了高糖誘導的p-Btk、 p-ERK、NF-κBp-p65和p-IκB表達(F=154.631,P<0.05;F=8.654,P<0.05;F=6 546,P<0.05;F=12.91,P<0.05)(圖7~9)。

圖6 高糖刺激在不同時間點對BMMs各蛋白表達的影響
2.8 PCI-32765對p65核轉移的影響為了研究PCI-32765在NF-κB信號路徑中的作用,使用激光共聚焦檢測p65核轉移。研究結果表明,在低糖組中,綠色熒光標記的NF-κB p65幾乎全部分布于細胞質;在高糖組中,綠色熒光主要分布在細胞核中。而PCI-32765抑制了高糖誘導的BMMs p65核轉移。見圖10。

圖7 PCI-32765對p-Btk表達的影響

圖8 PCI-32765對MAPK信號通路各蛋白表達的影響

圖9 PCI-32765對NF-κB信號通路蛋白表達的影響
3 討論
在中國慢性腎臟病透析前人群中,DN已經超過腎小球腎炎成為慢性腎臟病的主要病因,在不久的將來,DN將不可避免地成為導致透析的主要原因[9],研究其致病機制有助于減少透析人群,減輕社會經濟負擔。DN主要病理改變為巨噬細胞聚集、腎小球肥大、基底膜增厚、細胞外基質增加以及腎臟纖維化,最終引起腎功能損害。目前對于DN的研究[10]提示巨噬細胞浸潤程度與糖尿病的腎臟損害之間存在相關性。高糖導致巨噬細胞活化、增殖、分化為促炎性巨噬細胞,表達較高的iNOS,引起炎癥反應和組織改變。
越來越多的學者研究DN與慢性微炎癥的關系。本團隊曾經報道[11]過,炎癥介質如MCP-1和TNF-α,在糖尿病db/db小鼠的腎臟中表達增加。抑制炎性細胞因子的分泌可能延緩DN的進展。本研究結果表明,TNF-α、IL-1β和MCP-1的水平在高糖誘導的BMMs中增加,而高糖誘導的BMMs趨化較正常組織增加,提示高糖誘導巨噬細胞遷移的原因是上清中炎癥介質濃度增加,而Btk抑制劑PCI-32765,可以抑制MCP-1、TNF-α及IL-1β這些細胞炎癥因子的表達。這與高糖環境下iNOS表達激活巨噬細胞[12]、PCI-32765抑制巨噬細胞激活和炎癥反應結果一致。

圖10 PCI-32765對高糖刺激BMMs NF-κB p65核轉移的影響
高糖引起ERK信號通路在系膜細胞和足細胞中激活[13-14]。本研究表明,高糖可以激活巨噬細胞中ERK、JNK、p38MAPK以及NF-κB信號通路,PCI-32765抑制了高糖誘導的ERK和NF-κB信號通路激活,而p-JNK和p-p38的減少并不明顯,這進一步證實了Btk在ERK和NF-κB信號通路起到關鍵作用,同時在高糖誘導腎臟炎癥中發揮了重要作用。其中,NF-κB通過調節細胞炎癥因子的基因表達,參與腎臟炎癥反應。Yang et al[15]報道,抑制NF-κB通路可以抑制高糖刺激足細胞產生的炎性細胞因子。本研究結果提示,抑制Btk可以降低高糖誘導BMMs中TNF-α、 IL-1β和MCP-1水平。因此,PCI-32765可能通過抑制NF-κB信號通路,從而在抗炎中發揮作用;這與研究[4]提示的Btk調節巨噬細胞NF-κB磷酸化的結論相一致。
本研究結果表明,高糖通過調節ERK、 JNK、 p38MAPK以及NF-κB信號通路,誘導巨噬細胞的炎癥,參與DN發生。而Bruton酪氨酸激酶抑制劑PCI-32765,通過ERK及NF-κB信號通路降低了炎癥因子TNF-α、IL-1β以及MCP-1水平,提示Btk可能參與高糖環境下巨噬細胞通過ERK及NF-κB信號通路激活,參與調節高糖誘導巨噬細胞炎癥的發生,這為抑制DN炎癥反應提供了新的研究方向。
猜你喜歡
中國設備工程(2022年12期)2022-07-11 04:33:00
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2019年9期)2019-11-25 07:34:36
中學生數理化·七年級數學人教版(2019年9期)2019-11-25 07:34:34
中學生數理化·七年級數學人教版(2019年12期)2019-05-21 02:53:50
中學生數理化·七年級數學人教版(2019年12期)2019-05-21 02:53:48
