KLF2在肺動脈高壓大鼠肺組織中的表達及機制探討*
2020-09-09 06:49:14尚粉青葉玉蘭
陜西醫學雜志 2020年8期
王 蕊,尚粉青,葉玉蘭,郭 瑄
1.西安高新醫院心內科(西安710075);2.西安市第一醫院心內科(西安710002)
肺動脈高壓(Pulmonary hypertension,PAH)發病隱匿,診療棘手,預后差。近年來研究表明,肺血管內皮細胞功能紊亂是導致PAH肺血管異常收縮、肺血管重構和肺部血栓形成的主要原因[1]。Kruppel樣轉錄因子2(Kruppel-like factor2,KLF2)在肺組織高度表達,近來研究表明其在調節內皮細胞生物活性中起重要作用[2],因此我們預測其在PAH發生過程中可能起重要作用。本研究通過腹腔注射野百合堿(Monocrotaline,MCT)建立大鼠PAH模型,觀察KLF2及其相關因子表達變化,以期為進一步研究PAH干預措施提供理論依據。
材料與方法
1 實驗動物及材料 清潔級雄性SD大鼠24只,8~10周齡,體重(200±10)g,購于西安交通大學動物研究中心,飼養于西安交通大學醫學院心血管研究中心動物房;動物房為清潔級,動物的飼養條件:溫度(22±2)℃,標準12 h光照12 h黑暗節律,普通專用大鼠合成飼料喂養和飲水。野百合堿由購自Sigma公司。PCR引物由擎科生物技術有限公司合成。β-actin抗體購自Santa Cruz公司;eNOS抗體、KLF2、ET-1抗體購自美國Cell Signaling公司。NK-κB抗體、VEGFR2抗體購自美國Abcam公司。蛋白酶抑制劑及磷酸酶抑制劑(德國Roche公司),ECL發光液(Vazyme公司,Millipore公司),Trizol 試劑(Invitrogen公司),HiScriptTMqPCR Super Mix逆轉錄試劑盒(Vazyme公司),PCR DNA聚合酶(Vazyme公司),其余試劑為國產分析純。實時定量PCR儀及梯度PCR儀(美國Applied Biosystems 公司),WB電泳儀和電泳槽、WB半干轉膜槽(BIO-RAD公司),自動洗片機(柯達公司)。
2 動物模型的建立 所有實驗大鼠適應性飼養1周,隨機將其分為對照組(10只)、PAH模型組(MCT組,14只),均在同等條件下喂養。對照組按60 mg/kg腹腔注射0.9%氯化鈉溶液,MCT組按60 mg/kg腹腔一次性注射MCT造模。
3 血流動力學和心室肥厚檢測 MCT注射造模4 周后,稱量兩組大鼠體重 (BW),水合氯醛(5 ml/kg)腹腔注射麻醉,連接壓力換能器,將微導管 (PE50) 沿著大鼠頸外靜脈插入右心室,此時可見振幅較大的右心室波,并記錄右心室收縮壓(RVSP)。處死動物后取出心肺組織,去除心房及大血管,分離右心室和左心室以及室間隔,濾紙吸干組織并測量右心室質量(RV)、左心室質量(LV)和室間隔質量(S),計算右心室肥厚指數(RVHI)=右心室RV/(左心室LV+室間隔S)、右心室質量指數(RVMI) =RV/BW(mg/g)。肺組織標本一部分組織分成1 cm×1 cm×1 cm大小迅速存放于液氮中,后于-80 ℃凍存,用于分子生物學研究。剩余部分用4%多聚甲醛溶液固定。
4 血清各項指標測定 使用ELISA試劑盒測定血清中白介素-1β(IL-1β)、白介素-6(IL-6)及內皮素-1(ET-1)水平變化。采用硝酸還原酶法測定血清一氧化氮(NO)含量。
5 肺組織各項指標測定 ①取肺組織50 mg,剪碎,加入Trizol 1 ml超聲勻漿,提取組織RNA,反轉錄-聚合酶鏈反應(RT-PCR)方法測定肺組織中KLF2及核因子-κB(NK-κB)、血管內皮生長因子受體2(VEGFR2)、內皮素-1(ET-1)、一氧化氮合酶(eNOS)各因子的表達變化。PCR引物序列見表1。②取肺組織80 mg/只,剪碎組織,加入蛋白裂解液約300 ml,勻漿機勻漿,3次,每次30 s;液氮凍融3次,4℃,12 000 r/min,離心15 min,取上清,ABC法測定蛋白濃度;Western blot方法測定肺組織中上述各因子的蛋白表達的變化。

表1 RT-PCR引物列
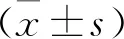
結 果
1 一般情況觀察及基本數據 MCT組在腹腔注射第2周開始出現毛發枯糙、喘息、食量減少,第25、26天分別有1只大鼠死亡,實驗結束時對照組無死亡。MCT組大鼠RVSP較正常對照組顯著增高(P<0.01),提示PAH模型制備成功(圖1A);正常對照組大鼠右心室形態正常,RVHI為(23.7±2.1)%,MCT組大鼠RVHI較正常對照組顯著增高(33.2±2.2)%(P<0.01)(圖1B);正常對照組大鼠RVMI為(0.81±0.15)%,MCT組大鼠RVMI較正常對照組顯著增高(1.25±0.24)%(P<0.01)(圖1C)。

注:與對照組相比,**P<0.01
2 兩組大鼠肺組織中KLF2及NK-κB、VEGFR2、ET-1、eNOS各因子表達比較 RT-PCR結果提示,MCT組大鼠肺組織中KLF2及eNOS的mRNA表達水平較對照組顯著降低;而NK-κB、VEGFR2及ET-1的mRNA表達水平較對照組顯著增高。Western blot結果提示,MCT組大鼠肺組織中KLF2及eNOS的蛋白表達水平較對照組顯著降低;而NK-κB、VEGFR2及ET-1的蛋白表達水平較對照組顯著增高。見圖2、3。

注:與對照組相比,**P<0.01 注:與對照組相比,*P<0.05,**P<0.01
3 兩組大鼠血清中IL-1β、IL-6及ET-1、NO水平測定結果 MCT組大鼠血清中IL-1β、IL-6及ET-1水平較對照組顯著升高,而NO水平較對照組顯著降低,見表2。
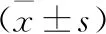
表2 兩組大鼠血清各因子表達情況
4 大鼠肺組織表達KLF2情況與血清各因子的相關性分析 見圖4。

圖4 大鼠肺組織KLF2與血清各因子相關性
Spearman相關性分析結果顯示:KLF2與血清IL-1β(r=-0.617,P=0.0006)(圖4A)、IL-6(r=-0.539,P=0.021)(圖4B)及ET-1(r=-0.881,P=0.006)(圖4C)呈顯著負相關,而與血清NO含量呈顯著正相關(r=0.721,P=0.001)(圖4D)。
討 論
肺動脈高壓指肺動脈壓異常升高的一種病理生理狀態,其發病機制復雜,尚未完全明確。目前治療主要集中在抑制肺血管平滑肌細胞增殖及肺血管收縮,波生坦作為一種ET-1受體拮抗劑應用于臨床取得了可喜的療效[3];西地那非為磷酸二酯酶V選擇性抑制劑,能增強NO釋放;外源性前列環素類似物,主要通過擴張肺動脈血管床[4]。亦有多種中藥顯示出一定的控制肺動脈高壓的作用[5-6]。上述藥物雖然可以改善生活質量,但是仍不能有效改善預后。因此需要進一步探討其發病上游更重要、更集中的因素所在,為進一步研究治療靶點提供理論依據。
目前有觀點表明,肺血管內皮細胞功能紊亂是PAH發生發展的基礎環節。KLF2作為KLF家族中重要的轉錄因子,在內皮功能調控中起重要作用。我們推測KLF2在PAH發病過程中可能有以下作用機制。①調節血管活性因子的生成:KLF2可抑制ET-1和ACE的表達,降低縮血管物質的生成[7];同時可直接結合到eNOS基因啟動子增加其轉錄活性[8],效誘導e NOS的表達,使血管NO生成增加。②越來越多的證據表明,炎癥反應在肺動脈高壓病理改變的形成中發揮關鍵作用[9-10];如炎癥因子IL-1β、IL-6、MCP-1及TNF-a均明顯增高[11];KLF2通過抑制炎性因子IL-1β、IL-18等的產生發揮抗炎作用[12]。研究表明[13]NF-κB在肺高壓大鼠肺組織表達顯著增高,參與肺高壓發展過程中氧化應激及炎癥反應的調控過程。KLF2可能與抑制NF-κB通路的多個組件相互作用,降低NF-κB的活性,間接發揮抗炎作用[14]。③血管增生重構:KLF2與VEGFR2啟動子結合抑制內皮細胞VEGFR2的表達[15],從而有抑制血管生成的作用;另有研究報道KLF2可抑制低氧誘導因子的表達,抑制低氧誘導的血管生成[7]。
本研究結果揭示PAH大鼠肺組織KLF2表達明顯減少。前期相關研究[16]亦揭示缺氧誘導的肺動脈高壓大鼠肺組織KLF2顯著降低,AMPK磷酸化ACE2后可誘導KLF2表達增加從而緩解肺高壓進展。肺動脈高壓縮血管物質ET-1表達顯著增加,而e NOS表達降低,NO生成減少,ET-1與NO均可被KLF2調控其表達,提示KLF2為多種血管活性物質的共同調節因子。同時,PAH大鼠肺組織NF-κB表達顯著增加,血清IL-1β表達亦明顯增高,提示炎癥因子表達明顯增高,KLF2可調節NF-κB的 表達及活性,從而對內皮細胞炎癥反應過程起到調控作用。由本研究結果可推測,KLF2在PAH發病發展過程中可能發揮關鍵作用,是多種血管活性物質生成的上游調節因素,是炎癥反應的上游調控因子,是血管重構的調節因素。因此,針對KLF2來進行PAH的治療干預措施的研究將是未來的方向之一。
猜你喜歡
體育科技文獻通報(2022年3期)2022-05-23 13:46:54
中老年保健(2021年3期)2021-08-22 06:50:04
天津外國語大學學報(2021年3期)2021-08-13 08:32:18
遼金歷史與考古(2021年0期)2021-07-29 01:06:54
昆明醫科大學學報(2021年1期)2021-02-07 01:06:36
現代臨床醫學(2021年1期)2021-01-26 00:56:02
中華養生保健(2020年4期)2020-11-16 01:31:40
中西醫結合肝病雜志(2020年2期)2020-10-27 02:18:50
科技傳播(2019年22期)2020-01-14 03:06:54
民用飛機設計與研究(2019年4期)2019-05-21 07:21:24
