碘海醇中間體及原料藥的合成研究
2020-09-12 14:16:33劉春李嘉駒代玉蘭
中國科技縱橫 2020年8期
劉春 李嘉駒 代玉蘭

摘 要:通過篩選催化劑,改進原料藥,合成碘海醇中間體及原料藥。目前從磷礦處理液中提取碘項目已取得成功,本方案通過篩選催化劑,改進原料藥,合成制備碘海醇中間體及其原料藥[1]。
關鍵詞:碘海醇;合成;原料藥
中圖分類號:TQ421.7 文獻標識碼:A 文章編號:1671-2064(2020)08-0216-02
1 現有技術路線分析
目前主要形成了三條的技術路線。
1.1 技術路線1
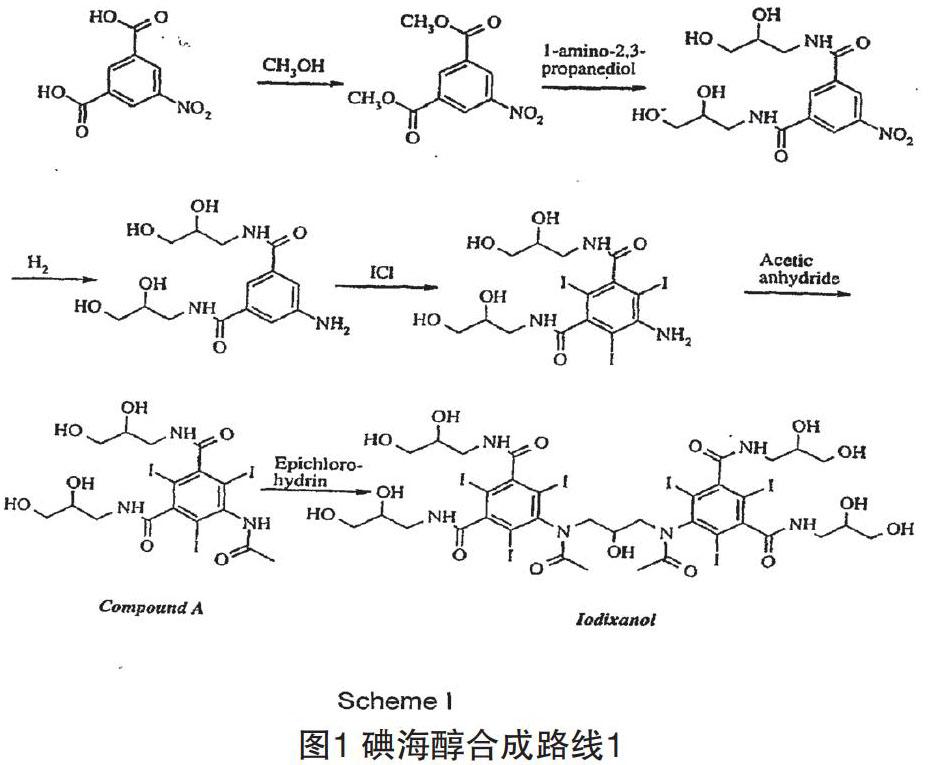
最早提出的技術路線是以5-硝基-1,3-二苯甲酸甲酯為原料,經酰胺化,還原,再酰化,最后N-烷基化而獲得碘海醇粗品(如圖1所示)。
這條路線的總收率較低,碘海醇原料藥的純化十分繁瑣和昂貴[2]。
1.2 技術路線2
該條路線中,第一步反應的的收率是65%,第二步與1-氨基-2,3-丙二醇酰化收率是80%,與3-氯-1,2-丙二醇進行N-烷基化的收率是54%。
本專利技術對這條路線的合成工藝進行了改進,提高了單步反應的收率。按路線(如圖2所示)合成碘海醇在技術和經濟上更占優勢[3]。
1.3 技術路線3
該技術路線(如圖3所示)和前兩條路線相比,收率有了較大提高,成本下降,操作安全性好[4]。
2 該項目采取的技術路線
2.1 合成碘海醇起始原料
技術路線3中所用的起始原料為5-氨基-2,4,6-三碘-1,3-二苯甲酰氯[5],但該原料價格昂貴,不利于降低碘海醇的制備成本,因此選用價格較低的5-硝基-1,3-二苯甲酸,經還原、碘化、酰氯化可以很方便得到起始原料5-氨基-2,4,6-三碘-1,3-二苯甲酰氯。
2.1.1 還原
該反應缺點是反應需在壓力反應器密閉進行,時間較長(24h),操作上具有一定危險性。因此用以下方式進行改進與優化:
該反應在常壓下進行,時間短(2h)左右,收率較高。
2.1.2 碘化
該反應的產率為80%左右。
2.1.3 合成二酰氯
該實驗中使用過量的SOCl2,既作試劑也作溶劑,過量的SOCl2和催化劑可以蒸餾回收用于下批次反應。常用的催化劑是DMF[6],但DMF也會產生以下缺點:(1)需升溫到90℃左右才能使反應完全,在此溫度下,催化劑會部分分解,并和底物反應生成不溶的、組成不確定的副產物。(2)有文獻報道DMF和SOCl2合用時,會形成有毒副產物(Levin, D. Org. Proc. Res. DeV. 1997, 1,182)。
因此,我們對催化劑進行優化,采用N-甲基吡咯烷酮或四甲基脲為催化劑,文獻中報道這兩種催化劑具有和DMF相似的催化活性、并可克服上述缺點。
2.2 碘海醇中間體Ⅰ、Ⅱ和原料藥的制備
獲得制備碘海醇的起始原料5-氨基-2,4,6-三碘基-1,
3-二苯甲酰氯后,采取以下路線來合成碘海醇中間體Ⅰ、Ⅱ和原料藥。
2.2.1 中間體Ⅰ和5-酰胺二酰氯的合成
實施方案A:100g 5-氨基-2,4,6-三碘-1,3-二苯甲酰氯懸浮在500g冰醋酸中,緩慢加入60gSOCl2,反應3h,冷卻后,所生成沉淀過濾回收,余液先用醋酸然后用水洗,母液中繼續產生的沉淀和第一次所生成沉淀合并,可獲得100g左右的產物,產率約93%(如圖4所示)。
實施方案B:采用乙酰氯代替冰醋酸做酰化試劑,反應在雙極性溶劑中進行,溶劑可選DMF,二甲基乙酰胺(DMA),二甲基亞砜(DMSO)或N-甲基吡咯烷酮。乙酰氯用量是二酰氯底物的1.5~3倍,反應時間24h以上,產物收集是將反應結束后的混合液滴入冰水中,沉淀出產物,過濾收集。反應產率和實施方案A相當。
2.2.2 中間體Ⅱ的合成
實施方案:370.4g5-乙酰-二酰氯(底物)溶于741.0gDMA,232.8g1-氨基-2,3-丙二醇溶于DMA,然后在0℃滴加入底物的DMA溶液中,兩小時滴完(如圖5所示)。反應在25℃反應7h進行完全。在真空度12mmHg,92℃蒸出DMA至干,冷卻到50℃,殘余物用950g甲醇和500g水溶解,混合物用7%NaOH溶液(質量濃度)調節pH值為10.5,并通過反復加入NaOH維持pH在10.5,使之形成5-乙酰氨基-N,N-雙(2,3-二羥丙基)-2,4,6-1,3-苯二甲酰胺鈉鹽。將產物沉淀和1-氨基-2,3-丙二醇的回收后,反應液冷卻到室溫,加入36%HCl調節pH值到1.0,沉淀產物,過濾收集,干燥。母液濃縮近干,加入500g水,透過2000mLNa型Amberjet1200型陽離子交換樹脂,1-氨基-2,3-丙二醇用7%氨水提取回收。
2.2.3碘海醇原料藥的制備
實施方案:5-乙酰氨基-2,4,6-三碘-N,N-雙(2,3-二羥丙基)-1,3-二苯甲酰胺,1,2-丙二醇為溶劑,加入甲醇鈉甲醇溶液,50℃攪拌溶解,減壓回收過量甲醇,冷卻至室溫,加入3-氯-1,2-丙二醇,室溫攪拌48h。反應結束后,減壓濃縮至干,冷卻,向剩余物加入甲醇,過濾,除去不溶物,向濾液中加水,陽離子交換樹脂,陰離子交換樹脂,攪拌2h,濾去樹脂,濾液減壓濃縮至干,冷卻,向剩余物加入正丁醇研磨,析出白色固體,得粗品,用正丁醇重結晶后的產率約82%(如圖6所示)。
技術風險及障礙:在進行N-烷基化步驟時,會伴隨O-烷基化,可能形成多種副產物,導致產物分離困難。
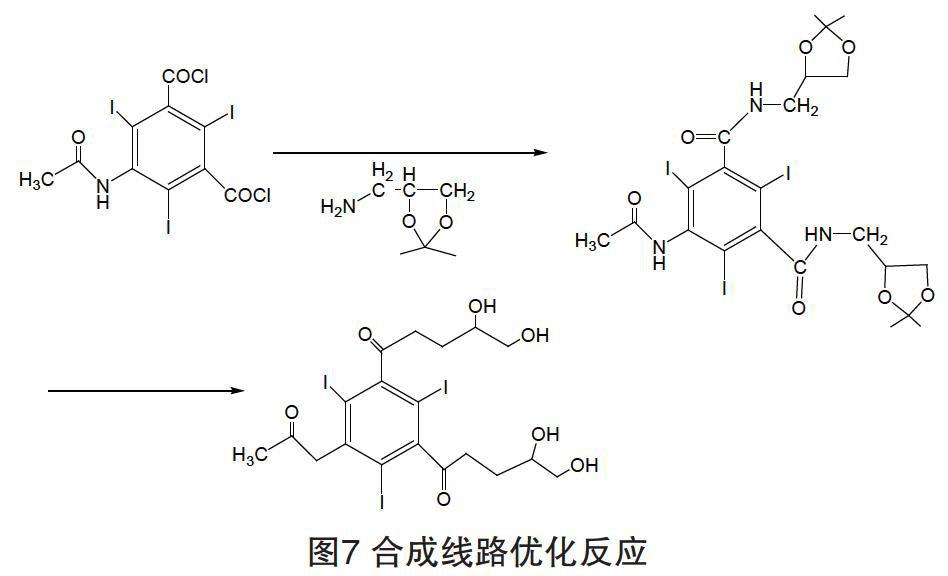
合成路線優化方案:在N-烷基化前在2,3位側鏈烷基上引入氨基丙縮酮,在烷基化步驟后用陽離子交換樹脂/甲醇-水體系脫去縮酮而獲得碘海醇原料藥(如圖7所示)。
優點:預防O-烷基化副產物的產生,后處理中用陽離子釋放H質子方式除去保護基,避免后處理中需采用重結晶來除去O-烷基化副產物。改進后反應時間縮短,操作簡便安全,成本降低,能耗少。
參考文獻
[1] 陳朝暉,董曉莉.離子色譜法測定食鹽中的碘含量[J].現代科學儀器,2004(5):65.
[2] 陳昭平,羅來濤.酸處理對海泡石表面及其結構性質的影響[J].南昌大學學報(自然科學版),2000,24(1):68-72.
[3] 陳兆能,邱澤麟,余經洪.試驗分析與設計[M].上海:上海交通大學出版社,1991.
[4] 黃憲.新編有機合成化學[M].北京:化工出版社,2003.
[5] 包丙男.近年來發展的非離子型水溶性有機碘造影劑[J].國外醫藥—合成藥、生化藥、制劑分冊,1986,7(5):275-277.
[6] 吳恩惠.介紹經腎排泄水溶性碘造影劑[J].中華放射性雜志,1992,26(2):131-132.
