食品中紅2G、二甲基黃、二乙基黃工業染料的同時測定
2020-09-18 07:14:26徐文泱趙紅清劉漾倫
食品與機械 2020年8期
關鍵詞:標準
梁 鋒 徐文泱 - 趙紅清 - 王 凱 劉漾倫 -
(1. 湖南省食品質量監督檢驗研究院,湖南 長沙 410017;2. 食品安全監測與預警湖南省重點實驗室,湖南 長沙 410017)

偶氮型工業染料的提取溶劑多為無機酸、甲醇、丙酮、乙腈、乙酸乙酯、水[3-4]等,試劑的種類應根據目標物的極性進行選擇;檢測采用的凈化手段有液液萃取[5]、固相萃取[6]、基質分散固相萃取[7]和凝膠滲透色譜技術[8]等。液相色譜及液相色譜質譜聯用法以其高選擇性和高靈敏度的特點,常被用來篩查和確證食品中的痕量工業染料[9-11]。Lim等[12]以C18SPE小柱對食品中的二甲基黃和二乙基黃凈化,結合液相色譜進行檢測,液相色譜質譜聯用儀確證,定量限分別達0.09,0.14 mg/kg。Yoshioka等[13]采用聚酰胺粉小柱實現飲料、糖漿和糖果基質樣品的凈化,光電二極管陣列液相色譜儀檢測水溶性色素紅2G,其定量限可達0.1 mg/kg。Minioti等[14]以水為溶劑,稀釋或超聲提取飲料、果醬、糖果中包括紅2G在內的13種合成染料,重點優化了流動相的選擇和洗脫程序。目前尚未建立這3種工業染料在食品中同時測定的檢驗方法,為了加強對這種在食品生產經營活動中可能出現的非法添加行為的監管,國家市場監管總局在2019年公開征集食品補充檢驗方法需求后,根據重要緊急以及科學可行的原則進行篩選,并組織專家評審將調味品、豆制品和肉制品中二甲基黃、二乙基黃和紅2G的檢測方法列為目前急需建立的食品補充檢驗方法,以加強對食品安全案件調查、食品安全事故處置等工作的技術支撐,提高監管效能。試驗擬建立3種工業染料在食品中的同時測定方法,采用液相色譜法定量,液相色譜—質譜聯用色譜儀進行定性確證,旨在為監測監督此類物質的非法使用提供依據。
1 試驗部分
1.1 儀器與設備
高效液相色譜儀:1260 Infinity型,美國Agilent公司;
液相色譜—質譜儀:Q Exactive Focus/U3000 Rs UPLC型,美國賽默飛世爾科技公司;
分析天平:CP214型,奧豪斯儀器(上海)有限公司;
旋轉蒸發儀:RE-2000A型,上海亞榮生化儀器廠。
1.2 材料與試劑
水:GB/T 6682規定的一級水;
鹽酸、無水硫酸鈉、乙酸銨:分析純,國藥集團化學試劑有限公司;
乙腈:色譜純,上海安譜實驗科技股份有限公司;
紅2G、二甲基黃、二乙基黃標準品:純度99.9%,北京壇墨質檢科技有限公司;
HLB固相萃取小柱:6 mL,300 mg,美國Waters公司。
1.3 樣品前處理
稱取5 g均質后的樣品于50 mL離心管中,加入15 mL 鹽酸酸化的50%乙腈—水溶液,渦旋混勻,4 500 r/min 離心2 min,重復提取一次后合并上清液,旋轉蒸發至近干,以50%乙腈—水溶液定容至2 mL,上清液待凈化。
HLB固相萃取小柱用3 mL乙腈活化,加入待凈化液,棄去流出液;加入3 mL乙腈,棄去流出液,加入4 mL 10%氨水—甲醇,收集流出液。將流出液于50 ℃下氮吹至干,用50%乙腈—水溶液定容至1 mL,過0.22 μm微孔濾膜,供高效液相色譜分析。如實際樣品中有檢出,通過保留時間及DAD光譜圖不能準確定性,可通過高效液相色譜—三重四極桿串聯質譜法進行確證。聚酰胺粉小柱、DMY固相萃取柱、PSA小柱的方法分別參照文獻[15-16]。
1.4 標準溶液配制
(1) 標準儲備液:分別稱取紅2G、二甲基黃、二乙基黃各0.010 0 g于100 mL容量瓶中,用50%乙腈—水溶液溶解并稀釋至刻度,搖勻,制成濃度為100 μg/mL的標準儲備液,0~4 ℃冷藏保存,有效期6個月。
(2) 混合標準中間液:分別準確吸取紅2G、二甲基黃、二乙基黃標準儲備液各5 mL于同一25 mL容量瓶中,用50%乙腈—水溶液稀釋至刻度,搖勻。0~4 ℃冷藏保存,有效期1個月。
(3) 混合標準工作液:分別準確吸取不同體積的混合標準中間液,用50%乙腈—水溶液稀釋成一系列標準工作液,臨用新制。
1.5 色譜條件
色譜柱:C18(2.1 mm×50 mm,1.8 μm);流動相:A為0.02 mol/L乙酸銨,B為乙腈,洗脫梯度程序見表1;流速1.0 mL/min;柱溫30 ℃;檢測波長:二甲基黃410 nm,二乙基黃418 nm,酸性紅507 nm;進樣量10 μL。

表1 色譜柱梯度洗脫程序
1.6 液相色譜—質譜條件
1.6.1 二甲基黃及二乙基黃液相色譜—串聯質譜/質譜測定
(1) 液相色譜條件:色譜柱為ACQUITY UPLC?BEH C1850 mm×2.1 mm,1.7 μm,進樣量5 μL;柱溫40 ℃;流速0.2 mL/min;流動相A為含0.1%甲酸+0.05 mol/L 乙酸銨—水溶液,流動相B為乙腈,按表2進行梯度洗脫。

表2 二甲基黃及二乙基黃液梯度洗脫程序
(2) 質譜條件:離子化方式為電噴霧電離;正離子掃描;多反應監測(MRM);噴霧電壓3 000 V;傳輸毛細管溫度500 ℃;MRM模式下參數設置見表3。

表3 多反應監測模式下二甲基黃、二乙基黃的質譜參數
1.6.2 紅2G液相色譜—串聯質譜/質譜測定
(1) 液相色譜條件:色譜柱為ACQUITY UPLC?BEH C1850 mm×2.1 mm,1.7 μm;進樣量5 μL;柱溫40 ℃;流速0.2 mL/min;流動相A為含0.05 mol/L乙酸銨—水溶液,流動相D為乙腈,按表4進行梯度洗脫。

表4 紅2G的梯度洗脫程序
(2) 質譜條件:離子化方式為電噴霧電離;負離子掃描;多反應監測(MRM);噴霧電壓2 500 V;傳輸毛細管溫度500 ℃;質譜參數設置見表5。

表5 多反應監測模式下紅2G的質譜參數
將標準系列工作液按液相色譜參考條件進行測定,測定相應的色譜峰面積,以標準工作液的濃度(μg/mL)為橫坐標,以色譜峰的峰面積為縱坐標,繪制標準曲線。按前述儀器條件測定樣品和混合標準系列工作溶液,記錄樣品和混合標準系列工作溶液中目標物的保留時間。若樣品中檢出與混合標準系列工作溶液中待測物保留時間一致的色譜峰(變化范圍在±2.5%內),且其定性離子與濃度相當的標準溶液中相應的定性離子的相對豐度相比偏差不超過表6規定的范圍,則可以確定樣品中檢出相應的待測物。

表6 定性確定時相對離子豐度的最大允許偏差
2 結果與討論
2.1 儀器條件優化
二甲基黃、二乙基黃呈堿性帶正電荷,以0.1%甲酸—水溶液和乙腈作為流動相峰形和信號靈敏度較好,紅2G可電離出H+,用乙酸銨或甲酸銨—水溶液作為流動相出峰峰形較好。采用液相色譜質譜聯用進行測定,當使用同一流動相時,3個化合物不能同時達到峰形良好的狀態。而液相色譜法采用0.02 mol/L乙酸銨和乙腈為流動相時,峰形良好,無雜質峰干擾,3個峰得到較好的分
離。因此選用液相色譜法作為定量方法,當液相色譜法無法進行準確定性時,采用液相色譜質譜聯用對其進行確證。由圖1~3可知,流動相中二甲基黃、二乙基黃、紅2G分別在410,418,507 nm處有最大吸收波長,因此分別選擇這3個波長為檢測波長。
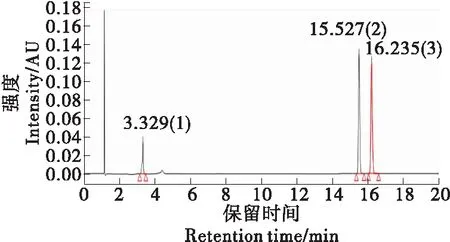
1. 紅2G 2. 二甲基黃 3. 二乙基黃
2.2 前處理條件優化
2.2.1 溶劑的選擇 配制的標準溶液澄清透明,靜置24 h 后無沉淀物或懸浮物,說明所選的溶劑對這3種色素的溶解性較好。
2.2.2 凈化柱的選擇 由圖4可知,聚酰胺粉對二甲基黃和二乙基黃的回收率不高,可能是由于pH為3~4時,其對色素的吸附效果最好;DMY的固相萃取小柱更適合于二甲基黃和二乙基黃的檢測,對于紅2G的凈化效果不理想;PSA固相萃取柱的平均回收率不高。HLB是一種親脂性與親水性填料混合的固相萃取柱[17-18],其準確性較其他3種凈化方式高。因此,在固相萃取柱的選擇方面采用具有親水性和疏水性的HLB固相萃取柱。

圖2 紅2G標準物質的總離子流色譜圖和提取離子色譜圖

圖3 二甲基黃和二乙基黃的標準物質的總離子流色譜圖和提取離子色譜圖

圖4 4種凈化方式對不同食品類別中3種偶氮性化合物回收率的影響
2.3 基質效應的測定
基質效應是以液相色譜—質譜聯用儀對復雜基質的痕量樣品進行檢測時應考慮的因素[19]。試驗方法的研究對象為調味品、肉制品和豆制品,其蛋白質和脂肪易影響凈化結果。選取3種不同的基質,其斜率比為80%~115%。因此可不考慮基質效應的影響。
2.4 線性范圍和檢出限
試驗表明,二甲基黃的線性回歸方程為Y=1.36×105x-7.03×103,相關系數r=0.999 8;二乙基黃的線性回歸方程為Y=1.49×105x-8.66×103,相關系數r=0.999 7;紅2G的線性回歸方程為Y=2.07×105x+9.18×103,相關系數r=0.999 7,表明在5.0~100.0 μg/mL內呈線性關系,符合定量要求。當稱樣量為5 g時,紅2G的檢出限為0.5 mg/kg,二甲基黃、二乙基黃的檢出限均為0.1 mg/kg。由此可知,采用此方法的檢出限低,3種偶氮型化合物的線性良好。
2.5 回收率和精密度
由表7可知,調制品中的回收率為81.2%~90.5%,肉制品中的回收率為80.7%~91.8%,豆制品中的回收率為81.4%~85.2%。試驗方法對加標食品中的3種偶氮型化合物檢測具有理想的回收率和精密度。

表7 辣椒醬中紅2G、二甲基黃、二乙基黃的回收率和精密度
3 結論
建立了同時測定調味品、肉制品及豆制品中紅2G、二甲基黃和二乙基黃等偶氮性工業染料的液相色譜法和LC-MS/MS方法。結果表明,試驗方法在0.1~10.0 μg/mL 內呈良好的線性關系,在添加濃度為0.000 5,0.001 0,0.005 0 mg/kg時的回收率為80.7%~91.8%,具有較為理想的回收率和精密度,滿足新方法確認的技術參數要求,為進一步強化檢驗檢測技術對食品安全監管的支撐作用提供了方法參考。后續可進一步增加偶氮型工業染料的數量,擴展研究的食品基質,建立多種工業染料的同時測定方法。
猜你喜歡
城市道橋與防洪(2022年4期)2022-07-01 06:04:12
當代陜西(2019年8期)2019-05-09 02:22:48
上海建材(2019年1期)2019-04-25 06:30:48
動漫星空(興趣百科)(2019年3期)2019-03-07 07:23:10
家庭影院技術(2018年4期)2018-05-09 07:07:52
專用汽車(2016年4期)2016-03-01 04:13:43
質量與標準化(2015年9期)2015-12-31 11:41:40
中國質量與標準導報(2014年4期)2014-03-11 19:54:25
中國質量與標準導報(2014年10期)2014-02-28 22:25:47
中國質量與標準導報(2014年7期)2014-02-28 22:24:39
