基于可逆離子氫鍵構筑室溫快速自修復聚硅氧烷彈性體
2020-09-25 08:36:10時金鳳符文鑫李志波
功能高分子學報 2020年5期
時金鳳, 趙 娜, 符文鑫, 李志波
(1.青島科技大學高分子科學與工程學院,山東 青島 266042;2.中國科學院化學研究所先進高分子材料實驗室,北京 100190)
聚硅氧烷彈性體是一類性能優異的材料,其生物相容性好,力學性能易調節,耐化學腐蝕性優異,在較寬的溫度范圍內擁有良好的彈性,被廣泛應用于柔性電子器件[1]、耐高低溫密封材料[2]、人造血管[3]等方面。對比傳統的熱固性彈性體材料,自修復材料具有修復損傷的性能,從而延長了產品的使用壽命,某種程度上能夠減少材料的浪費[4]。
近年來,許多文獻報道了賦予材料自修復功能的不同方法,從而拓展了其在涂料[5]、黏合劑[6]、傳感器[7]、柔性電子器件[8]等領域的應用。制備自修復型聚硅氧烷彈性體的方法主要有:(1)植入微膠囊[9],首先制備能夠包覆催化劑的微膠囊,再將微膠囊埋入材料基體。當材料受外力作用發生破壞時,應力誘導微膠囊破裂,催化劑從膠囊中釋放,分散在材料基體中的預聚體在催化劑作用下發生化學反應,進而實現斷裂處自修復。但此時微膠囊的尺寸和分散性等會影響彈性體最終的力學性能,且不能實現多次自修復。(2)引入可逆共價鍵,如在聚合物基體中引入亞胺鍵[10]、雙硫鍵[11]、Diels-Alder 反應形成可逆的化學鍵[12,13],以及硼酸酯或硼氧六環鍵[14,15]等。可逆共價鍵在保證材料力學性能的基礎上賦予材料自愈合性能,同時也避免了植入微膠囊引入的化學污染。但實現可逆共價鍵通常需要外界條件的充分刺激,比如長時間或深層次的光、熱刺激等,并且需要相對復雜的單體分子結構設計和合成。(3)引入非共價鍵,如氫鍵[16]、離子鍵[17]、金屬配位鍵[18-20]等。與前兩種方式相比,非共價鍵的鍵能較弱,易于斷裂與重組,當材料出現斷裂缺口時,更易通過非共價鍵的重組實現自修復。另外,利用可逆共價鍵和非共價鍵或多種類型非共價鍵的協同作用,可以同時提高材料的力學強度和可拉伸性能。例如,Zhang 等[21]報道了含有脲基和雙硫鍵交聯的聚硅氧烷彈性體,體系中大量的脲鍵形成分子鏈內/間的氫鍵弱相互作用,當材料發生大形變時進行能量耗散,從而賦予材料優異的柔韌性;另外,可逆的雙硫鍵可維持聚合物網絡的完整性,提高材料的力學強度。Kang 等[22]將6-苯基-2,2’-二吡啶官能團引入聚硅氧烷主鏈,利用環二價鉑金屬絡合物之間Pt-Pt 和π-π 兩種非共價相互作用形成交聯網絡,制備的彈性體表現出優異的柔韌性;但是多種化學鍵的引入也增加了材料設計、合成以及性能調控的難度。
通常,未補強的聚硅氧烷彈性體的斷裂伸長率小于300%[23],而且這類材料是基于硅氫加成反應實現共價交聯的聚硅氧烷彈性體,并不可回收和自修復。因此,探索簡單高效的方法制備高度可拉伸、可回收、可快速自修復的聚硅氧烷彈性體材料對于拓寬聚硅氧烷材料在下一代智能材料領域(如高分子驅動器、軟體機器人、電子皮膚等)的應用具有重要意義。
基于上述研究背景,本文通過實驗室自制的環三聚磷腈堿(CTPB)[24,25]作為催化劑,在溫和條件下高效合成了具有不同分子量和硅氧乙烯基單元摩爾分數(χ,連接有C=C 雙鍵的Si―O 單元占全部Si―O 單元的比例)的線型聚硅氧烷前驅體,并通過硫醇-烯點擊化學反應,簡單高效地制備了側基分別為羧基和氨基的聚硅氧烷(PDMS-g-COOH 和PDMS-g-NH2)。將兩種聚合物溶液充分均勻混合,成功制備了由羧基與氨基間離子氫鍵交聯的聚硅氧烷彈性體(PDMS-g-[COOH/NH2]),進而研究了聚合物基體中乙烯基含量、聚合物分子量對彈性體拉伸性能、自修復性能的影響。結果表明,PDMS-g-[COOH/NH2]彈性體具有良好的伸展性能,且在室溫條件下可以實現快速自修復。本研究為制備可低能量驅動自修復的聚硅氧烷彈性體提供了一種簡單通用的制備方法。
1 實驗部分
1.1 原料和試劑
八甲基環四硅氧烷(D4)、四甲基四乙烯基環四硅氧烷(V4):純度98.0%,上海阿拉丁生化科技股份有限公司,使用前加入氫化鈣常溫下攪拌24 h,減壓蒸餾,并收集在盛有分子篩的存儲瓶密封保存;封端劑四甲基二乙烯基硅氧烷(純度97%)、巰基丙酸(純度99%):上海阿拉丁生化科技股份有限公司;CTPB:實驗室自制;巰基乙胺鹽酸鹽(純度98%)、光引發劑2,2-二甲氧基-苯基苯乙酮(DMPA,純度99%):安耐吉有限公司;甲苯:分析純,煙臺遠東精細化工有限公司,使用前經有機純化系統純化除水;四氫呋喃、甲醇:分析純,天津富宇精細化工有限公司。
1.2 測試與表征
凝膠滲透色譜(GPC)儀(美國,Agilent 1260):以單分散聚苯乙烯(PS)為標樣,測試溫度為40 ℃,THF 為流動相,流速為1.0 mL/min;紅外光譜(FT-IR)儀(德國,Bruker Tensor 27):測試波長范圍600~4 000 cm-1,采用衰減全反射(ATR)模式;核磁共振波譜(NMR)儀(瑞士,Bruker AVANCE NEO 400 MHz):溶劑為氘代氯仿(CDCl3)、氘代甲醇(CD3OD)、氚代水(HOD);拉力試驗機(美國,Instron 5900):拉伸速率分別為50 、100 、200 mm/min,試驗樣條長為4 cm,寬為3 mm。
1.3 實驗步驟
1.3.1 甲基乙烯基線型聚硅氧烷(PDMS-g-Vi)的合成 首先,稱取24 mg(0.02 mmol)CTPB 置于單口瓶中并加入1 mL 甲苯充分攪拌溶解,在另外一個史萊克瓶加入計量好的D4、V4、封端劑(具體配比如表1 所示)和適量甲苯充分溶解。然后,將CTPB 的甲苯溶液轉入上述單體混合溶液中,30 ℃下攪拌反應30 min 后,將反應液緩慢倒入50 mL 甲醇中沉降析出聚合物,用甲醇充分洗滌3 次,去除小分子和寡聚物。最后,于真空烘箱中室溫真空干燥得到目標聚合物PDMS-g-Vi,其合成路線如圖1(a)所示。根據調整投料比(n(D4)/n(V4))與聚合物在體系中的濃度(如表1 所示),得到了3 種線型聚硅氧烷前驅體(P1,P2,P3)。

表1 CTPB 催化D4 和V4 開環共聚合結果Table 1 Ring-opening polymerization of D4 and V4 catalyzed by CTPB
1.3.2 PDMS-g-COOH 的合成 以P1-g-COOH 為例:稱取2.5 g P1(5 mmol 乙烯基)和50 mL THF 置于100 mL史萊克瓶中,氮氣鼓泡排除體系中的空氣。在氮氣保護下,將25 mg(0.1 mmol)光引發劑DMPA 和1.6 g(15 mmol)巰基丙酸加入到上述溶液中混合均勻(巰基丙酸與乙烯基的摩爾比為3),室溫條件下,汞燈光照30 min 后,將聚合物的THF 溶液倒入去離子水中沉降,重復水洗3 遍,除去多余的巰基丙酸,室溫下真空干燥得到P1-g-COOH。通過1H-NMR 確定接枝率大于99%,其合成過程如圖1(b)所示。
1.3.3 P1-g-NH2的合成 稱取2.5 g P1(5 mmol 乙烯基)和30 mL THF 置于100 mL 史萊克瓶中,氮氣鼓泡排除體系中的空氣。在氮氣保護下,將25 mg(0.1 mmol)光引發劑DMPA、1.7 g(15 mmol)巰基乙胺鹽酸鹽的甲醇(6 mL)溶液加入到上述溶液中混合均勻(巰基丙酸與乙烯基的物質的量之比為3),室溫條件下,汞燈光照30 min 后,聚合物的THF 溶液用飽和碳酸鈉水溶液中和,倒入去離子水中沉降,重復水洗3 次,除去多余的巰基乙胺鹽酸鹽,室溫下真空干燥得產物P1-g-NH2。通過1H-NMR 確定接枝率大于99%,其合成過程如圖1(b)所示。
1.3.4 PDMS-g-[COOH/NH2]彈性體的制備 以P1-g-[COOH/NH2]彈性體為例:將等物質的量的P1-g-COOH與P1-g-NH2分別溶于THF 中,質量濃度約為10 mg/mL。將P1-g-COOH 的THF 溶液逐滴加入到PDMS-g-NH2的THF 溶液中,攪拌過夜。將得到的黏稠聚合物溶液倒入四氟乙烯模具中,室溫過夜,溶劑自然揮發后,放入真空干燥箱進一步除去溶劑,得到P1-g-[COOH/NH2]彈性體。采用相同的方法將P2-g-COOH、P3-g-COOH 分別與P1-g-NH2制備得到另外兩種聚合物彈性體,命名為P2-g-[COOH/NH2]彈性體和P3-g-[COOH/NH2]彈性體。樣品的制備過程如圖1(c)所示。
2 結果與討論
2.1 PDMS-g-Vi 的表征
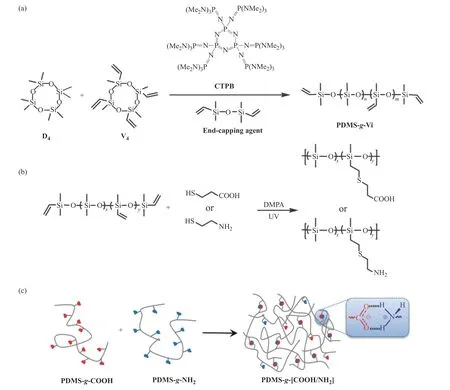
圖1 (a)PDMS-g-Vi,(b)P1-g-COOH 與P1-g-NH2,(c)PDMS-g-[COOH/NH2]的合成示意圖Fig.1 Synthetic routes of (a)PDMS-g-Vi,(b)P1-g-COOH and P1-g-NH2,(c)PDMS-g-[COOH/NH2]
PDMS-g-Vi 的數均分子量(Mn)、分子量分布(?)通過GPC 表征,χ 通過核磁共振氫譜表征,結果如表1所示。一般由堿性陰離子引發的環硅氧烷開環聚合最終會達到平衡,得到的聚合物中混有大量的線型寡聚物和不同分子量的環硅氧烷中間體,導致聚合物的分子量分布較寬(?>2)[26,27]。然而用CTPB 催化開環聚合的結果顯示出了高單體轉化率(>80%)和相對較窄的分子量分布(?<2),符合非平衡陰離子開環聚合的特點[28]。此外,當投料比n(D4)/n(V4)=85/15 時,通過調整聚合物在體系中的濃度,由1.5 mol/L 增加到2.5 mol/L,可以成功制備乙烯基摩爾分數相似,數均分子量分別為43.0×103和144.8×103的P1 和P2。通過調節n(D4)/n(V4)由85/15 增加至92/8,保持聚合物在體系中的濃度為2.5 mol/L,制備的P2 和P3 其Mn相近,但χ 分別為14.5%和7.1%。
2.2 改性聚硅氧烷及其彈性體的結構表征
圖2 是P1、P1-g-COOH 和P1-g-NH2的1H-NMR 譜圖。在聚硅氧烷前驅體P1 的1H-NMR 譜圖中,化學位移5.70~6.05(b, c)處的多重峰歸屬于乙烯側基(CH2=CH―)的氫原子信號。經巰基丙酸改性以后的P1-g―COOH,在0.85(e)、2.47~2.70(g, f)和2.70~2.84(h)處出現的新峰分別歸屬于3種亞甲基(―Si―CH2―,―CH2―S―CH2―,COOH―CH2―)上氫的化學位移;同時,乙烯側基在5.70~6.05(b, c)處的峰也完全消失,說明了P1-g-COOH 的成功合成。P1-g-NH2的結果與P1-g-COOH 的結果類似,與P1 對比,0.85(i)、2.49~2.66(k, j)、2.78~2.92(l)處出現的新信號峰分別歸屬于―Si―CH2―、―CH2―S―CH2―、NH2―CH2―的化學位移,并且乙烯側基的信號峰完全消失,也證明了硫醇-烯加成反應的完成。另外,P2、P3及羧基改性的產物P2-g-COOH 和P3-g-COOH 也顯示出類似的結果。
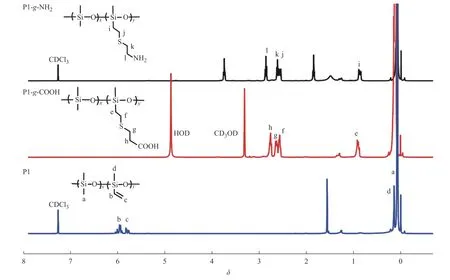
圖2 樣品的1H-NMR 譜圖Fig.2 1H-NMR spectra of samples
P1、 P1-g-COOH 和 P1-g-NH2的 FT-IR 結 果(圖(3))也證實了硫醇-烯加成反應的成功。如圖3所示,前驅體P1 在1 598 cm-1和3 054 cm-1處分別顯示出乙烯側基C=C 和C―H 的伸縮振動特征吸收峰[29,30]。經過巰基丙酸(紅線)改性后,P1-g-COOH在1 714 cm-1處出現新的吸收峰,歸屬于引入的羧基官能團C=O 的伸縮振動峰[29],并且在1 598 cm-1和3 054 cm-1處C=C 雙鍵上的特征吸收峰消失,進一步說明了P1-g-COOH 的成功合成,與圖2 結果吻合。同時,巰基乙胺(藍線)改性后的P1-g-NH2的FT-IR 譜圖于1 588 cm-1處顯示寬吸收峰,歸屬于引入氨基的N―H 彎曲振動[31],也證明了P1-g-NH2成功合成。此外,交聯彈性體P1-g-[COOH/NH2]的FTIR 結果顯示C=O 在1 714 cm-1處伸縮振動吸收峰右移,與對應N―H 在1 572 cm-1處的彎曲振動峰形成新的寬吸收峰,充分說明COOH 與NH2之間存在相互作用形成了離子氫鍵。聚合物P2 和P3、羧基改性產物P2-g-COOH 和P3-g-COOH,以及其與P1-g-NH2形成的彈性體的FT-IR 結果也顯示出類似的結果。
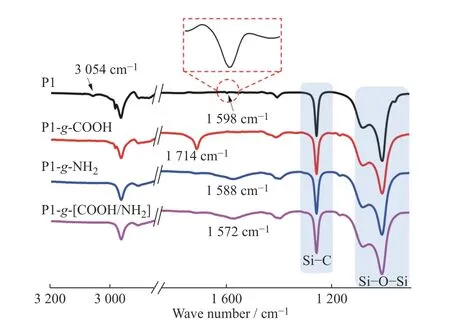
圖3 樣品的FT-IR 譜圖Fig.3 FT-IR spectra of samples
2.3 PDMS-g-[COOH/NH2]彈性體的力學性能
當體系中存在大量動態結合的離子氫鍵時,氫鍵可為材料提供可觀的力學性能和優異的延展性。作為一種中等或強氫鍵,離子氫鍵的鍵能為5~35 kJ/mol,仍遠低于共價鍵和離子鍵,屬于非共價弱鍵相互作用[32]。PDMS-g-[COOH/NH2]彈性體的交聯網絡正是基于羧基和氨基的離子氫鍵,如圖1(c)所示。
圖4 為P1-g-[COOH/NH2]彈性體在不同拉伸速率下的應力-應變曲線。由圖可知,P1-g-[COOH/NH2]彈性體的力學性能對拉伸速率有很強的依賴性。隨著拉伸速率的增加,彈性體的最大伸長率降低。在低拉伸速率(50 mm/min)條件下,P1-g-[COOH/NH2]彈性體中聚硅氧烷主鏈可以緩慢松弛,同時分子鏈間離子氫鍵的斷裂速率與重組速率相匹配,即斷裂的離子氫鍵具有足夠的時間完成重組,彈性體的交聯網絡仍舊得以保持,表現出較好的延伸性,其斷裂伸長率可達到877.0%。當拉伸速率依次增大到100 mm/min 和200 mm/min 時,其對應的最大斷裂伸長率分別降低到733.4%和522.3%。這是因為分子鏈在短時間內產生很大滑移,由拉伸應力所產生的部分斷裂的離子氫鍵難以實現重組,致使樣品斷裂。另外,根據時溫等效原理,隨拉伸速率的增加,分子鏈達到相應應變所需的時間縮短,短時間尺度高分子網絡的力學松弛行為表現出類似于熱固性材料的彈性行為,所以拉伸斷裂強度也逐漸增加,斷裂伸長率也隨之降低。
圖5 為PDMS-g-COOH 與P1-g-NH2所形成的彈性體在50 mm/min 拉伸速率下的應力-應變曲線。結果顯示:在相同的拉伸速率下,通過改變聚合物的分子量、離子氫鍵的交聯密度,可以調整所得彈性體的拉伸性能。在χ 接近的情況下(表1 中的P1 和P2),相應彈性體的拉伸強度從230.9 kPa 增加到344.5 kPa,同時斷裂伸長率也從877.0%增加到1 038.1%。聚硅氧烷鏈段分子量增加,分子鏈間的纏結程度增大,從而在一定程度上限制了分子鏈的滑移,提高了材料的力學強度;同時,在交聯密度相近的條件下,分子量越大的聚合物,其延展性能越好。另外,當聚合物數均分子量相近時,隨著χ 從P3 的7.1%增加到P2 的14.5%,相應彈性體的拉伸強度由231.9 kPa 提高到344.5 kPa,說明增加聚合物分子鏈間的離子氫鍵交聯密度能夠增強彈性體網絡的剛性,提高聚合物的拉伸強度,但柔韌性下降,斷裂伸長率由1 394.7%降低到1 038.1%。
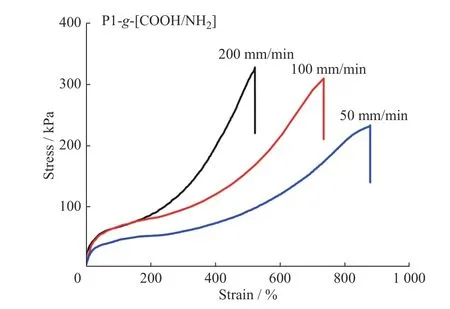
圖4 P1-g-[COOH/NH2]彈性體在不同拉伸速率下的應力-應變曲線Fig.4 Tensile stress-strain curves of P1-g-[COOH/NH2] elastomer at different stretching speeds

圖5 PDMS-g-[COOH/NH2]彈性體在 拉伸速 率50 mm/min條件下的應力-應變曲線Fig.5 Tensile stress-strain curves of PDMS-g-[COOH/NH2]elastomers at the stretching speed of 50 mm/min
2.4 PDMS-g-[COOH/NH2]彈性體的自修復性能
文獻中報道基于可逆共價鍵的自愈合彈性體大多需要外界輔助條件(如:高溫[21]、UV[10]等)才可以進行自修復,且修復效率依賴于外界能量輸入。因此,對低能量驅動的自修復材料的研究具有重要應用價值。基于離子氫鍵的弱相互作用,其相對更容易的動態成鍵/斷鍵為構筑可室溫快速自修復材料提供了一種可能。以P1-g-[COOH/NH2]彈性體為例,本文對其在室溫下的自修復性能進行了研究。如圖6(a)所示,首先將樣品裁成兩段,將兩段樣條在切口處對齊并緊貼在一起后,在室溫下分別靜置15 min 和30 min,無其他外部刺激,然后測其拉伸性能,結果如圖6(b)所示。
將修復后樣條的斷裂強度與原始樣品斷裂強度的比值定義為彈性體的修復效率。從應力-應變結果可以看出,P1-g-[COOH/NH2]彈性體的自修復過程非常快。靜置15 min 后,修復效率可以達到79%;靜置30 min后,達到99%,且拉伸強度完全恢復到原始樣條的拉伸強度,說明P1-g-[COOH/NH2]彈性體的自修復效率隨時間的增加而增加。P1-g-[COOH/NH2]彈性體的快速自修復性能歸因于構成網絡結構的COO-/NH3+離子氫鍵在受到外力時動態可逆的快速成鍵,同時也得益于主鏈Si―O―Si 鍵良好的柔順性,使得側基官能團有較好的結合效率。當把P1-g-[COOH/NH2]彈性體的斷裂面處重新組合在一起時,游離態的COO-與NH3+基團能夠非常迅速地重組,形成新的離子氫鍵,從而實現材料的自修復。
3 結 論
(1)利用CTPB 催化D4與V4開環共聚制備不同數均分子量和不同硅氧乙烯基單元摩爾分數的線型聚硅氧烷,進而通過硫醇-烯點擊化學反應,簡單高效地成功合成側基羧基或氨基改性的聚硅氧烷PDMS-g-COOH、PDMS-g-NH2,最后利用側鏈上羧基與氨基之間的離子氫鍵交聯形成自修復彈性體PDMS-g-[COOH/NH2] 。
(2)PDMS-g-[COOH/NH2]彈性體的力學強度和拉伸性能可通過改變聚合物的分子量、離子氫鍵交聯密度來調節。
(3)此類離子氫鍵交聯的聚硅氧烷彈性體在室溫下具有快速自修復性。斷裂樣品在室溫下靜置愈合30 min后,其拉伸強度和斷裂伸長率可完全恢復到原始樣品的相應水平。
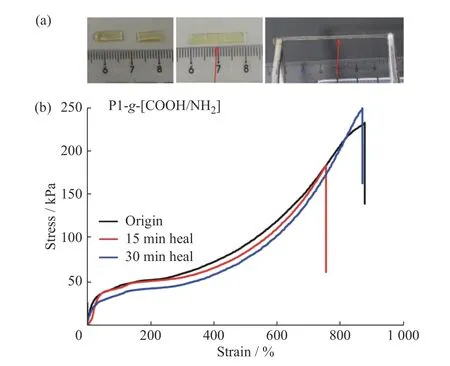
圖6 (a)P1-g-[COOH/NH2]彈性體室溫自修復過程;(b)樣品修復前后的應力-應變曲線(拉伸速率為50 mm/min)Fig.6 (a)Self-healing behavior of P1-g-[COOH/NH2] elastomer at room temperature;(b)Tensile stress-strain curves of P1-g-[COOH/NH2] elastomer before and after healing(the stretching speed was 50 mm/min)
