氮雜環(huán)丙烷參與的1,3-偶極環(huán)加成反應(yīng)研究進(jìn)展
2020-10-24 08:41:00趙森
福建質(zhì)量管理 2020年19期
關(guān)鍵詞:催化劑
趙 森
(荊州職業(yè)技術(shù)學(xué)院 湖北 荊州 434020)
氮雜環(huán)丙烷的參與的環(huán)加成反應(yīng)是在許多生物活性天然產(chǎn)物和藥物中獲得作為核心結(jié)構(gòu)骨架的各種含氮雜環(huán)的有力策略。適用于各種含氮環(huán)狀化合物的合成,因?yàn)樗苋菀鬃鳛?,3-偶極合成子與各種親偶極物反應(yīng),氮雜環(huán)丙烷的高反應(yīng)性歸因于它易于形成1,3-偶極子由路易斯酸介導(dǎo)的中間體。氮雜環(huán)丙烷是最小的環(huán)狀應(yīng)變含N雜環(huán)化合物,在有機(jī)合成中得到了廣泛的研究。
一、路易斯酸催化的氮雜環(huán)丙烷環(huán)加成反應(yīng)
(一)路易斯酸催化的氮雜環(huán)丙烷環(huán)加成反應(yīng)合成咪唑類化合物
1991年,Jin-Ook Baeg和Howard Alper報(bào)道了,雙(芐腈)二氯化鈀是氮雜環(huán)丙烷與碳二亞胺的環(huán)加成反應(yīng)形成咪唑啉亞胺的有效催化劑,產(chǎn)率為40-94%[1]。該方法是區(qū)域特異性的,涉及更多取代的環(huán)碳-氮鍵的裂解。其中一個(gè)咪唑啉基亞胺的X射線結(jié)構(gòu)確定,以及所有環(huán)加成反應(yīng)產(chǎn)物的光譜和分析數(shù)據(jù),為結(jié)構(gòu)分配提供了基礎(chǔ)。
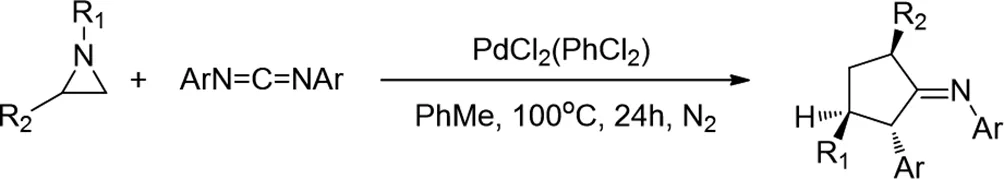
圖1
2017年,Pinaki Bhusan De等人報(bào)道了銅催化的2-烷基-/2-芳基氮雜環(huán)丙烷與苯并咪唑的交叉偶聯(lián)[2]。該反應(yīng)涉及氮雜環(huán)丙烷與苯并咪唑的區(qū)域特異性開環(huán),得到苯并咪唑基乙胺衍生物,其導(dǎo)致C(sp2)-H和N-H鍵之間的脫氫交叉偶聯(lián),產(chǎn)生二氫咪唑并咪唑。光學(xué)活性的2-芳基氮雜環(huán)丙烷可以立體可逆地交叉偶聯(lián),具有高對映體純度(77-97%ee)。這些需氧催化體系在中等溫度下包含廉價(jià)的Cu(II)鹽和PCy3配體。
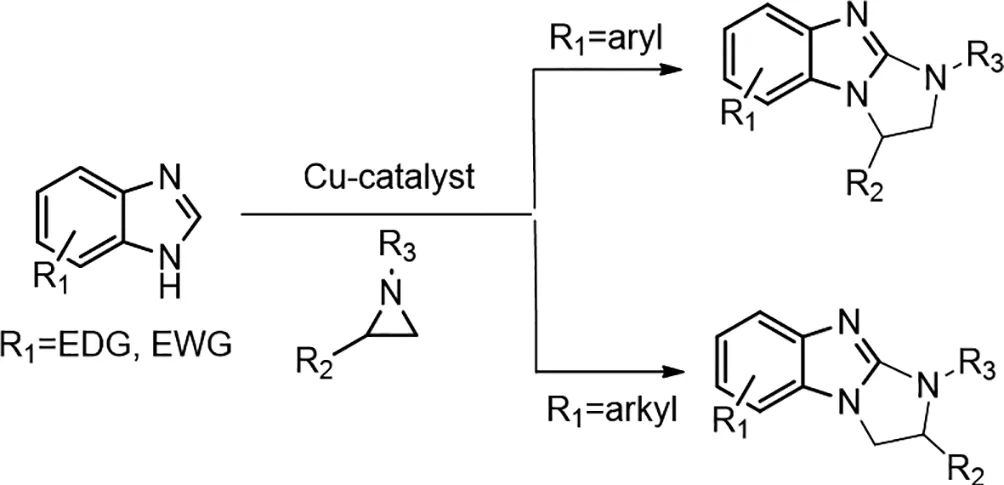
圖2
(二)路易斯酸催化的氮雜環(huán)丙烷環(huán)加成反應(yīng)合成吡咯類化合物
2009年P(guān)aul A.Wender和Daniel Strand報(bào)道了由路易斯(AgSbF6)或布朗斯臺德酸催化的氮雜環(huán)丙烷和非活化的炔之間的正式[3+2]環(huán)加成反應(yīng),得到了2,3-二氫吡咯[3]。該反應(yīng)溫和條件下進(jìn)行,可擴(kuò)展多種底物,使用緩慢的催化劑負(fù)載,高度區(qū)域選擇性,得到提供多種多取代的二氫吡咯。

圖3
2009年,Jinmin Fan等人通過使用FeCl3催化的氮雜環(huán)丙烷與芳基炔的反應(yīng),構(gòu)建了一系列新的高官能化的2-吡咯啉,其中涉及芳基取代的鏈烯基陽離子[4]。

圖4
2010年,Matthew Brichacek等人報(bào)道了銅催化劑催化的手性乙烯基氮雜環(huán)丙烷可以立體特異性地環(huán)擴(kuò)展的反應(yīng)[5]。該合成方法以具有適當(dāng)幾何形狀的氮雜環(huán)丙烷作為起始材料,控制獲得手性2,5-順式-或2,5-反式-3-吡咯啉產(chǎn)物。得到了二十三個(gè)環(huán)擴(kuò)展實(shí)例,其中大多數(shù)具有立體特異性環(huán)化。

圖5
2015年Linqing Wang等人通過原位產(chǎn)生的鎂催化劑介導(dǎo)內(nèi)消旋-氮雜環(huán)丙烷和C3-烷基吲哚之間的不對稱形式[3+2]-環(huán)加成,合成不對稱吡咯并吲哚啉的衍生物。通過在易于制備的非手性配體的幫助下使用市售的配體,可以獲得多種吡咯并吲哚啉[6]。

圖6
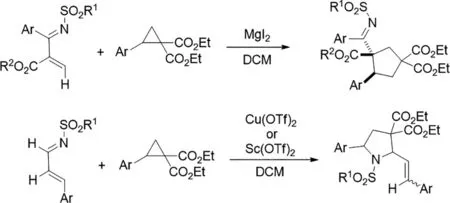
圖7
2018年,Pei-Jun Yang等人提出了第一路易斯酸([(CH3CN)4Cu]PF6)催化的外消旋2-(雜)芳基-N-磺酰基氮雜環(huán)丙烷經(jīng)C-N鍵與親核試劑裂解的立體聚合轉(zhuǎn)化[8]。這包括[異]芳香醛和1,3-二取代吲哚的[3+2]環(huán)加成反應(yīng),與富電子(雜)芳烴的不對稱Friedel-Crafts型反應(yīng),以及與胺的不對稱氨解反應(yīng),為手性1,3-異惡唑烷,吡咯并吲哚啉,2-(雜)芳基苯乙胺和鄰位二胺的合成提供了可靠途徑。基于對照實(shí)驗(yàn)的結(jié)果,提出了涉及外消旋氮雜環(huán)丙烷的I型動(dòng)態(tài)動(dòng)力學(xué)不對稱轉(zhuǎn)化(DyKAT)的機(jī)理。

圖8
2018年,Mateo Alajarin等人通過相應(yīng)的Yb(OTf)3和Y(OTf)3產(chǎn)生的N-芳基-C,C-二苯基乙烯亞胺與金屬羰基和金屬-甲亞胺葉立德的有效[3+2]環(huán)化促進(jìn)了碳-碳鍵雜化[9]。已經(jīng)完成了供體-受體環(huán)氧乙烷和氮雜環(huán)丙烷在溫和條件下進(jìn)行反應(yīng),得到了結(jié)構(gòu)上復(fù)雜的惡唑烷和吡咯烷衍生物,這是一種區(qū)域選擇性構(gòu)建該化合物的方法。此外,加熱N-芳基-C,C-二苯基乙烯亞胺和2,3-二羧酸二乙基己酯的混合物得到咪唑烷衍生物。通過計(jì)算研究這些[3+2]環(huán)加成的反應(yīng)機(jī)理。
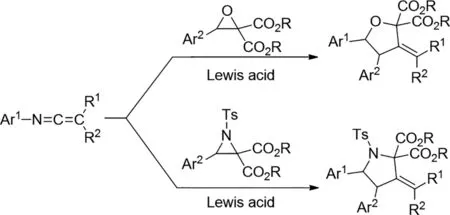
圖9
(三)路易斯酸催化的氮雜環(huán)丙烷環(huán)加成反應(yīng)合成苯胺類化合物
2014年,Ke Gao等人發(fā)現(xiàn)發(fā)現(xiàn)鈷-N-雜環(huán)卡賓催化劑通過開環(huán)烷基化促進(jìn)2-芳基吡啶與1,2-二芳基氮雜環(huán)丙烷的鄰C-H官能化[10]。該反應(yīng)在溫和的室溫條件下產(chǎn)率良好,得到了帶有2-氨基官能團(tuán)的1,1-二-芳基甲烷。
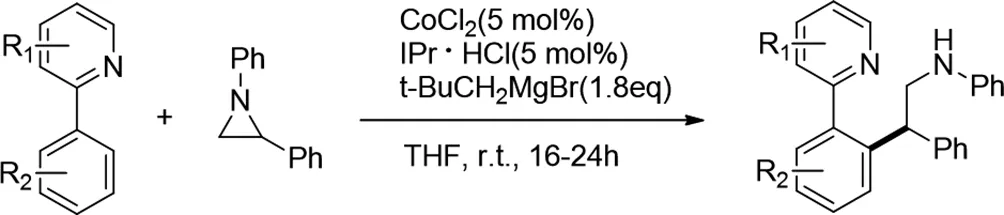
圖10
(四)路易斯酸催化的氮雜環(huán)丙烷環(huán)加成反應(yīng)合成惡唑類化合物
1995年,Jin-Ook Baeg等人發(fā)現(xiàn)了含有適當(dāng)取代基的1,2,3-三取代的氮雜環(huán)丙烷,可以與雜環(huán)戊烯在Pd(II)的催化下反應(yīng),得到五元環(huán)雜環(huán)[11]。該反應(yīng)具有較高的區(qū)域選擇性和立體特異性,保留了氮雜環(huán)丙烷環(huán)中取代基的立體化學(xué)結(jié)構(gòu)。1,2-二取代的氮雜環(huán)丙烷與雜環(huán)戊烯反應(yīng),得到相應(yīng)的手性五元環(huán)雜環(huán),反應(yīng)在保持構(gòu)型的情況下進(jìn)行。

圖11
2005年,Takeshi Munegumi等人發(fā)現(xiàn),在鎳催化劑存在下,氮雜環(huán)丙烷與異氰酸酯進(jìn)行環(huán)加成反應(yīng),并分離出五種亞氨基惡唑烷衍生物[12]。在NiI2存在下反應(yīng)時(shí)最佳。加長反應(yīng)的時(shí)間,NiI2催化亞氨基惡唑烷異構(gòu)化,生成相應(yīng)的咪唑烷酮衍生物。

圖12
2017年,Xingxing Wu等人報(bào)導(dǎo)了由Ni(II)-二惡唑啉絡(luò)合物催化的N-甲苯磺酰亞胺和醛的非對映和對映選擇性形式[3+2]環(huán)加成反應(yīng)。獲得的1,3-惡唑烷產(chǎn)物具有高非對映選擇性,良好的收率(高達(dá)99%)[13]。通過手性轉(zhuǎn)移方法實(shí)現(xiàn)具有挑戰(zhàn)性的遠(yuǎn)距離立體聲中心控制。
換油周期的延長,對潤滑油的剪切穩(wěn)定性提出了挑戰(zhàn)。此次展會(huì),阿朗新科還將介紹其針對這一市場變化推出的Keltan OCP極強(qiáng)穩(wěn)定性的牌號K0500R。該牌號是無定形OCP黏指劑,具有優(yōu)異的低溫性能,其剪切穩(wěn)定指數(shù)(SSI)為18,符合當(dāng)前市場的最新技術(shù)發(fā)展要求。目前,阿朗新科正在積極與潤滑油行業(yè)內(nèi)的標(biāo)桿企業(yè)選用K0500R開展長壽命柴油機(jī)油行車實(shí)驗(yàn)。
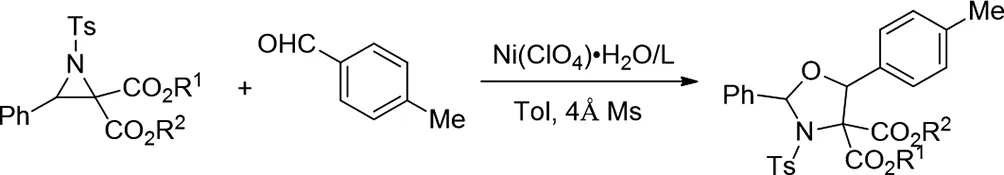
圖13
2017年,Sunatda Arayachukiat等人使用過渡金屬配位化合物作為路易斯酸和親核助催化劑的雙催化體系,開發(fā)了從相應(yīng)的N-甲苯磺酰基嘧啶和CO2區(qū)域選擇性合成N-甲苯磺唑烷酮的有效方法[14]。在篩選的路易斯酸時(shí),無鹵素的五乙醇鈮(Nb(OEt)5在四丁基碘化銨(TBAI)存在下顯示出最佳的催化活性。由催化實(shí)驗(yàn)支持的系統(tǒng)DFT計(jì)算表明,CO2插入是該過程的速率決定步驟,并且它高度依賴于鈮中心的空間位阻。

圖14
(五)路易斯酸催化的氮雜環(huán)丙烷環(huán)加成反應(yīng)合成惡嗪類化合物
2018年,Honglei Liu等人報(bào)道了Ni-催化的[8+3]環(huán)加成的庚三烯酮與2-芳基-N-甲苯磺酰胺在溫和的反應(yīng)條件下順利進(jìn)行,得到各種4-甲苯磺酰基-2,3,4,4a-四氫環(huán)庚[b]-[1,4]惡嗪衍生物,產(chǎn)率適中至優(yōu)異[15]。
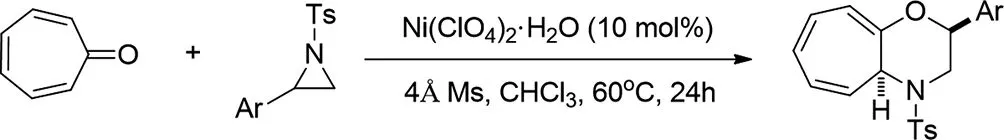
圖15
2018年,Abhijit Mal等人通過路易斯酸催化的Sn2型開環(huán)活化的氮雜環(huán)丙烷與2-鹵代苯酚,然后在一鍋條件下以逐步方式進(jìn)行Cu(I)催化的分子內(nèi)C-N環(huán)化,得到3,4-二氫-1,4-苯并惡嗪衍生物,產(chǎn)率高(高達(dá)95%)[16]。該策略為(S)-3甲基-1,4-苯并惡嗪(S)-3v提供短而有效的合成,后者是左氧氟沙星合成中的后期中間體。該方法有效且簡單,具有優(yōu)異的對映和非對映特異性(ee>99%,de>99%)。
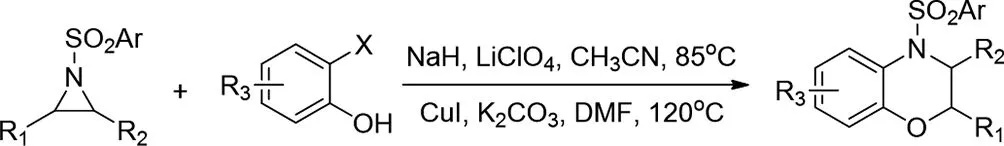
圖16
(六)路易斯酸催化的氮雜環(huán)丙烷環(huán)加成反應(yīng)合成惡二嗪類化合物
2015年,Stalin R.Pathipati等人報(bào)道了高選擇性路易斯酸催化的三元雜環(huán)與硝酮的環(huán)加成反應(yīng)[17]。使用Al或In催化劑,使用環(huán)氧乙烷,氮雜環(huán)丙烷和硫雜環(huán)丁烷作為底物用于合成各種六元雜環(huán)。該催化方案顯示出廣泛的底物范圍,并且在溫和的反應(yīng)條件下以高產(chǎn)率和優(yōu)異的選擇性提供了獲得新結(jié)構(gòu)基序的途徑。
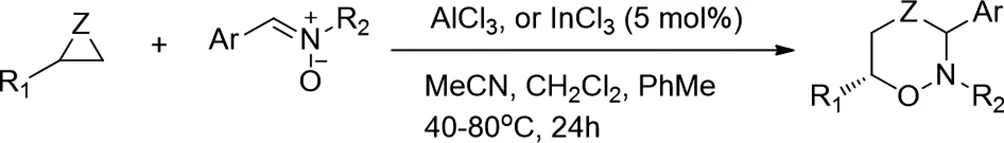
圖17
(七)路易斯酸催化的氮雜環(huán)丙烷環(huán)加成反應(yīng)合成噠嗪類化合物
2018年,Alexey O.Chagarovskiy等人報(bào)道了兩種不同三元環(huán)的(3+3)-環(huán)化的第一個(gè)實(shí)例。發(fā)現(xiàn)與二氮雜環(huán)丙烷反應(yīng)的供體-受體環(huán)丙烷在溫和路易斯酸(Ni)催化下以高產(chǎn)率和非對映選擇性提供全氫噠嗪衍生物[18]。該反應(yīng)適用的底物范圍廣泛,并表現(xiàn)出優(yōu)異的官能團(tuán)耐受性。
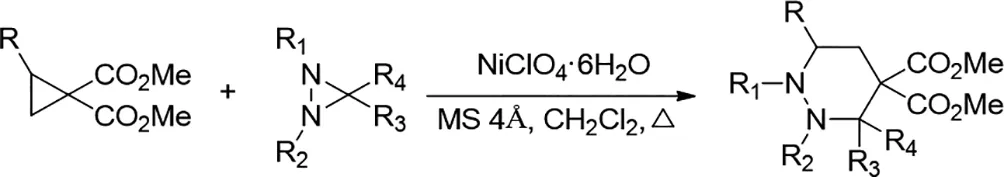
圖18
(八)路易斯酸催化的氮雜環(huán)丙烷環(huán)加成反應(yīng)合成吡嗪類化合物
2018年,Bijay Ketan Das等人報(bào)道了,具有炔丙基胺的立體特異性Cu催化的N-磺酰基氮雜環(huán)丙烷開環(huán),親核加氫胺化得到哌嗪,雙鍵異構(gòu)化以提供四氫吡嗪[19]。光學(xué)活性的氮雜環(huán)丙烷可以與高對映體純度(>98%ee)交叉偶聯(lián)。

圖19
(九)路易斯酸催化的氮雜環(huán)丙烷環(huán)加成反應(yīng)合成氮雜卓類化合物
2014年,Ming-Bo Zhou開發(fā)了一種合成氮雜環(huán)庚烷衍生物(一種典型的七元雜環(huán)系統(tǒng))的實(shí)用方法,該方法涉及使用六氟銻酸催化氮雜環(huán)丙烷與兩個(gè)炔的正式[3+2+2]環(huán)加成反應(yīng)[20]。該方法適用于兩種相同或不同的末端炔烴,用于[3+2+2]環(huán)加成與未活化的氮雜環(huán)丙烷,并提供相應(yīng)的吖庚因衍生物,產(chǎn)率高,具有良好的化學(xué)和區(qū)域選擇性。
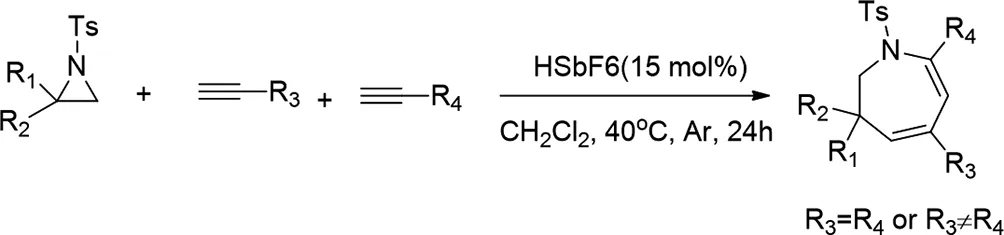
圖20
(十)路易斯酸催化的氮雜環(huán)丙烷環(huán)加成反應(yīng)合成哌嗪類化合物
2012年,Piera Trinchera等人報(bào)道了通過在催化量的路易斯酸(MgBr2)使未活化的N-烷基芳基氮雜環(huán)丙烷反應(yīng),直接合成順式和反式2,5-二取代的N,N-二烷基哌嗪對映體的反應(yīng)。并經(jīng)立體化學(xué)和核磁共振研究揭示這一過程的機(jī)制[21]。

圖21
(十一)路易斯酸催化的氮雜環(huán)丙烷環(huán)加成反應(yīng)合成噻唑烷類化合物
2016年,Mani Sengoden等人報(bào)道了,Al(salen)Cl有效地催化未活化的手性氮雜環(huán)丙烷與異硫氰酸酯的對映特異性(3+2)環(huán)加成,在室溫下提供94-99%ee的官能化亞氨基噻唑烷。使用鋁路易斯酸作為催化劑,高對映體純度,溫和的反應(yīng)條件,寬的底物范圍和高原子經(jīng)濟(jì)性是重要的實(shí)用特征[22]。

圖22
(十二)路易斯酸催化的氮雜環(huán)丙烷環(huán)加成反應(yīng)合成噻嗪類化合物
2017年,Rohit Kumar Varshnaya和Prabal Banerjee報(bào)道了路易斯酸(MgI2)催化N-甲苯磺酰基二羧酸酯和環(huán)氧乙烷的[3+3]環(huán)化,原位生成的巰基醛用于合成官能化的噻嗪和氧雜噻吩衍生物的方法[23]。另外,該方法通過去甲基化和Krapcho單羧化促進(jìn)噻嗪的衍生化。
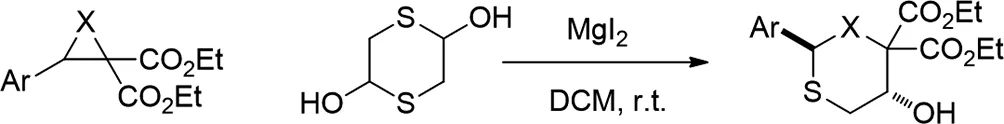
圖23
2018年,Murugan Vijay等人用1,4-二噻烷-2,5-二醇制備了N-磺酰基氮雜環(huán)丙烷的多米諾雙催化C-N/C-S鍵,在室溫下得到3,4-二氫-1,4-噻嗪[24]。使用Bi(OTf)3作為催化劑,原子經(jīng)濟(jì)性和區(qū)域選擇性是重要的實(shí)用特征。
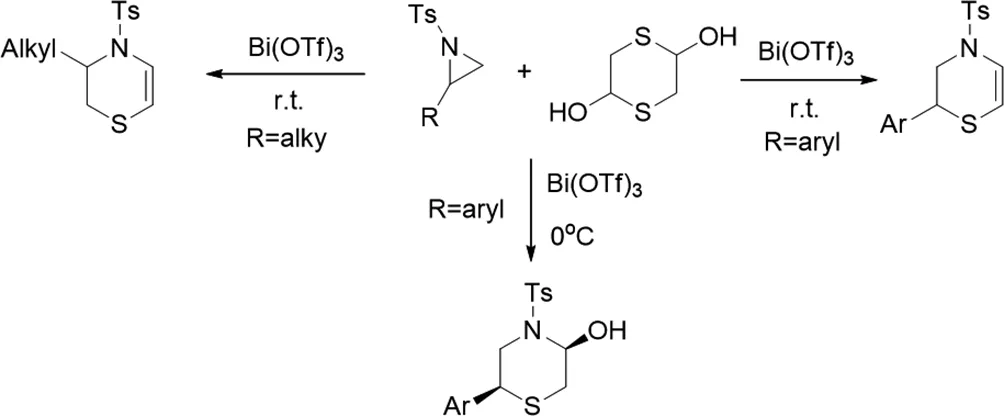
圖24
(十三)路易斯酸催化的氮雜環(huán)丙烷環(huán)加成反應(yīng)合成異喹啉類化合物
2018年,Sang Gyu Lee和Sung-Gon Kim報(bào)道了供體受體氮雜環(huán)丙烷與N,N-二烷基-3-乙烯基苯胺的路易斯酸催化的[3+3]環(huán)加成反應(yīng),用于立體選擇性合成四氫異喹啉(THIQ)[25]。使用Gd(OTf)3作為路易斯酸催化劑進(jìn)行的反應(yīng)對各種N-甲苯磺酰氮雜環(huán)丙烷和N,N-二烷基-3-乙烯基苯胺底物具有耐受性,并且通常以高產(chǎn)率獲得高官能化THIQ,具有好的非對映選擇性。
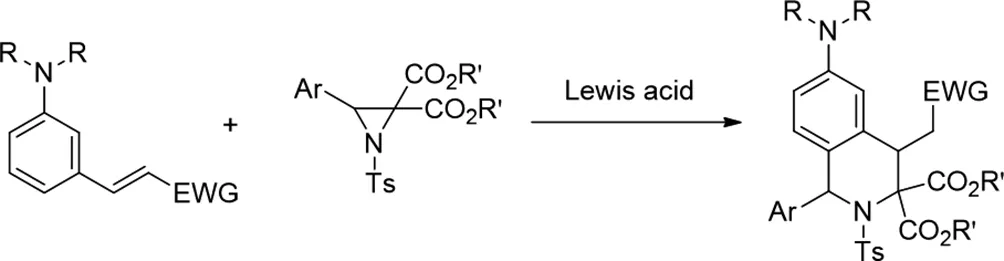
圖25
2019年Seungyeon Kim和Sung-Gon Kim使用Mg(OTf)2/雙惡唑啉催化劑建立了供體-受體氮雜環(huán)丙烷與N,N-二烷基-3-乙烯基苯胺的非對映體和對映體選擇性[3+3]環(huán)加成反應(yīng)[26],得到了高官能化的四氫異喹啉。各種N,N-二烷基-3-乙烯基苯胺,如m-N,N-二甲基氨基苯基α,β-不飽和苯基酮和3-(mN,N-二甲基氨基苯基)丙烯酸酯,以及供體受體N-甲苯磺酰基嘧啶,已應(yīng)用于這種不對稱催化方案。
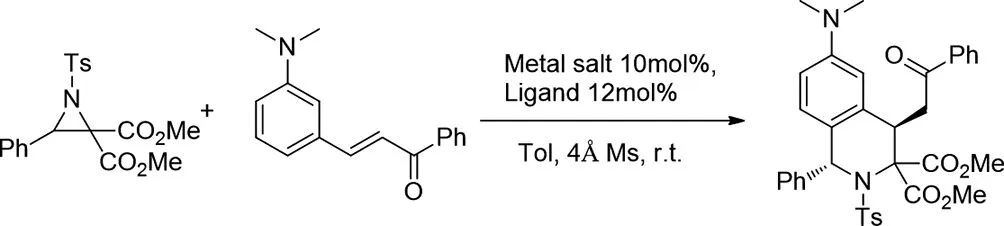
圖26
(十四)路易斯酸催化的氮雜環(huán)丙烷環(huán)加成反應(yīng)合成咔啉類化合物
2017年,Masthanvali Sayyad等人報(bào)道了,通過激活的氮雜環(huán)丙烷開環(huán)化(DROC)與2-乙烯基吲哚環(huán)合成具有優(yōu)異立體選擇性(de,ee至>99%)的各種1,4-二取代四氫-β-咔啉衍生物[27]。該反應(yīng)通過LiClO4催化的Friedel-Crafts型烷基化2-乙烯基吲哚與活化的氮雜環(huán)丙烷,然后以多米諾骨牌方式進(jìn)行分子內(nèi)氮雜-邁克爾反應(yīng)。
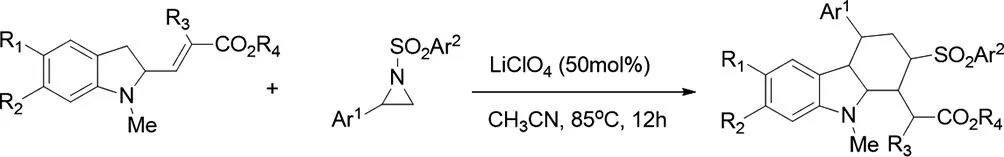
圖27
(十五)路易斯酸催化的氮雜環(huán)丙烷環(huán)加成反應(yīng)合成哌啶類化合物
2017年,Yizhou Zhan等人報(bào)道了在氮雜環(huán)丙烷2,2-二酯和共軛二烯之間的新型路易斯酸(三氟甲磺酸鈧)催化的[3+2]分子內(nèi)交叉環(huán)加成(IMCC)[28]。這是第一個(gè)具有碳=碳雙鍵的偶氮甲堿葉立德分子內(nèi)1,3-偶極環(huán)加成的區(qū)域特異性IMCC,并為構(gòu)建結(jié)構(gòu)復(fù)雜和多樣化的氮雜[n.2.1]骨架提供了一種通用且有效的策略。[3+2]IMCC可在溫和條件下以克規(guī)模進(jìn)行。更重要的是,3-烷基取代的氮雜環(huán)丙烷也是成功的。優(yōu)異的結(jié)構(gòu)多樣性,簡便的操作和多樣化的后修飾將支持[3+2]IMCC在天然產(chǎn)物合成和藥物發(fā)現(xiàn)中的應(yīng)用。
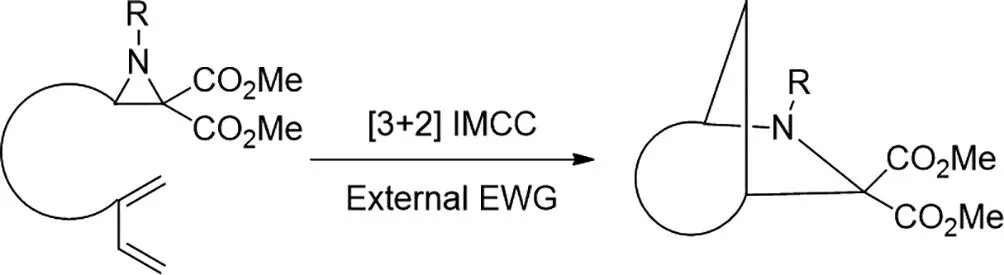
圖28
二、金屬卟啉配合物催化的氮雜環(huán)丙烷的偶極環(huán)加成反應(yīng)
2017年,Xun Wang等人報(bào)道了一種新的多孔金屬-金屬卟啉骨架MMPF-10,由一種八角形卟啉配體構(gòu)成,該配體連接銅槳輪單元,形成具有強(qiáng)滲透力的拓?fù)浣Y(jié)構(gòu)的骨架[29]。卟啉配體的原位金屬化為MMPF-10提供了兩個(gè)獨(dú)特的可接近的Cu(II)中心。這使得它在首次報(bào)道的CO2與氮雜環(huán)丙烷的反應(yīng)中表現(xiàn)為有效的路易斯酸催化劑,以合成由MMPF催化的惡唑烷酮。

圖29
2019年,Satoru Teranishi等人報(bào)道了有效的鐵卟啉路易斯酸催化的氮雜環(huán)丙烷與醛的環(huán)加成反應(yīng)[30],以提供具有高區(qū)域和非對映選擇性的惡唑烷。該反應(yīng)在25℃下,甲苯中與1mol%的鐵催化劑一起進(jìn)行。基于理論研究和基于同步加速器的X射線吸收精細(xì)結(jié)構(gòu)測量提供了對氮雜環(huán)丙烷-鐵卟啉絡(luò)合物的基本見解,該絡(luò)合物是產(chǎn)生1,3-偶極合成子的關(guān)鍵中間體。
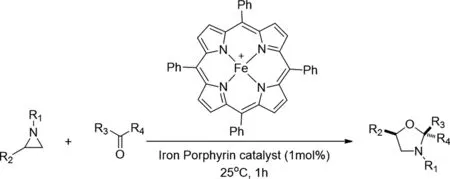
圖30
三、熱反應(yīng)導(dǎo)致氮雜環(huán)丙烷開環(huán)發(fā)生的環(huán)加成反應(yīng)
氮雜環(huán)丙烷的骨架結(jié)構(gòu)為鍵角接近60o的三元環(huán),這種結(jié)構(gòu)的環(huán)張力非常大,在加熱的條件下即可開環(huán),得到甲亞胺葉立得離子,從而與其他化合物進(jìn)行反應(yīng)。很多科學(xué)家基于這種理論對氮雜環(huán)丙烷的環(huán)加成反應(yīng)進(jìn)行了探索。
(一)熱反應(yīng)氮雜環(huán)丙烷環(huán)加成合成吡咯類化合物
1986年,Wayne K.Anderson等人報(bào)道了由甲硅烷基亞胺鹽或氮雜環(huán)丙烷或2H-氮雜環(huán)丙烷經(jīng)1,3-偶極環(huán)加成反應(yīng),合成了一系列雙[(氨基甲酰氧基)甲基]吡咯啉衍生物[31]。將吡咯啉的抗腫瘤活性與相應(yīng)的吡咯進(jìn)行比較。除C-2-偕二甲基取代的吡咯啉外,其他幾種衍生物均具有抗癌活性。
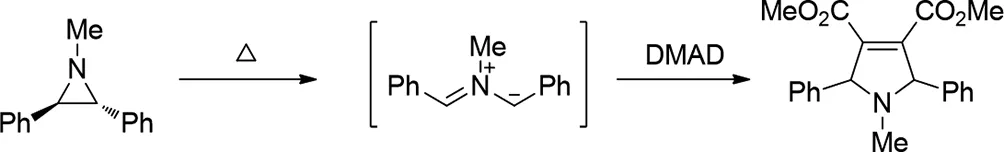
圖31
2014年,Alexander F.Khlebnikov等人報(bào)道了,量子化學(xué)計(jì)算1-甲基-2,3-二苯基-和1,2,3-三苯基氮雜環(huán)丙烷的熱開環(huán),形成相應(yīng)的S-,U-和W-型甲亞胺葉立德以及使用PCM溶劑化模型,在DFT B3LYP/6-31G(d)理論水平下進(jìn)行它們對乙炔二甲酸二甲酯(DMAD)和2,3-二氰基-2-丁二酸二Jinmin Fan甲酯的環(huán)加成反應(yīng)。計(jì)算完全符合實(shí)驗(yàn)結(jié)果,并解釋了根據(jù)親偶極子和內(nèi)鎓鹽中的取代基從協(xié)同路徑到非協(xié)同路徑的轉(zhuǎn)換。發(fā)現(xiàn)在親偶極物中,例如在二氰基丁二酸二烷基酯中,強(qiáng)吸電子取代基顯著降低了偶氮甲堿葉立德與這種偶極子反應(yīng)中形成兩性離子中間體的阻隔性。這可以使逐步循環(huán)加載與協(xié)同循環(huán)加速競爭。然而,甚至對具有幾個(gè)吸電子取代基的親偶極物的環(huán)加成的一致性受到葉立德和親偶極物對應(yīng)物中電子和空間效應(yīng)的精細(xì)平衡的支配。假設(shè)在偶氮甲炔葉立德中引入取代基使相應(yīng)兩性離子中的正電荷不穩(wěn)定將有利于協(xié)同環(huán)加成,即使用二烷基二氰基丁二酸酯也在理論上和實(shí)驗(yàn)上進(jìn)行了測試[32]。
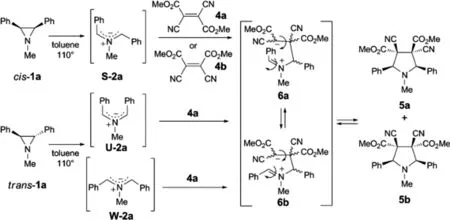
圖32
(二)熱反應(yīng)氮雜環(huán)丙烷環(huán)加成合成噻唑烷類化合物
2009年,Grzegorz Mloston′等人報(bào)道了,反式-1-甲基-2,3-二苯基氮雜環(huán)丙烷(反式-1a)與芳族和環(huán)脂族硫代酮2,在沸騰的甲苯中,得到相應(yīng)的順式-2,4-二苯基-1,3-噻唑烷順式-4,該反應(yīng)是通過反式-1a的開環(huán)旋轉(zhuǎn)得到中間體(E,E)-配位的甲亞胺葉立德3a的[2+3]-環(huán)加成反應(yīng)(方案1)。順式-1a與乙炔二羧酸二甲酯(5)的類似反應(yīng)得到反式-2,5-二氫-1-甲基2,5-二苯基吡咯-3,4-二甲酸酯(反式-6)。另外,順式-1a和反式-1a與二氰基富馬酸二甲酯(7a)以及順式-1a和二氰基馬來酸二甲酯(7b)的反應(yīng)產(chǎn)生相同的兩種立體異構(gòu)體二甲基3,4-二氰基的混合物。3,4-二氰基-1-甲基-2,5-二苯基吡咯烷-3,4-二羧酸酯8a和8b。相反,順式-1,2,3-三苯基氮雜環(huán)丙烷(順式-1b)和7a僅得到一個(gè)立體異構(gòu)的吡咯烷-3,4-二羧酸酯10,具有基于軌道對稱性控制預(yù)期的構(gòu)型,即通過協(xié)同反應(yīng)步驟[33]。
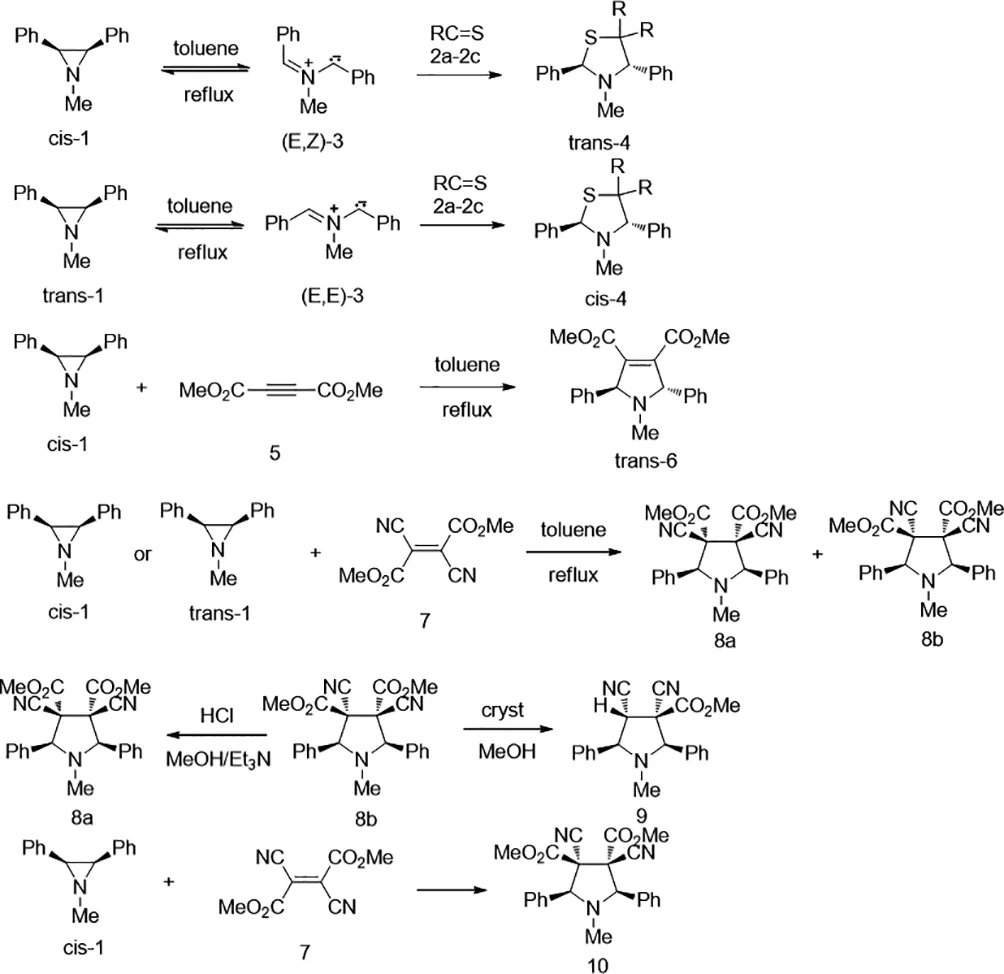
圖33
(三)熱反應(yīng)氮雜環(huán)丙烷環(huán)加成合成1,3-二氧戊環(huán)-2-酮類化合物
2011年,Hui Zhou等人報(bào)道了使用3-巰基丙基三甲氧基硅烷作為硅烷偶聯(lián)劑,使1,3-雙-(4-烯丙基-2,6-二異丙基苯基)咪唑氯化物與MCM-41反應(yīng),合成N-雜環(huán)卡賓(NHC)官能化MCM-41及其CO2。通過與CO2的反應(yīng)進(jìn)一步合成2加合物(稱為MCM-41-IPr-CO2)。原位漫反射紅外傅立葉變換光譜(DRIFTS)用于研究MCM-41-NHC的可逆CO2捕獲釋放能力。MCM-41-IPr-CO2加合物被證明是一種有效的非均相催化劑,用于將CO2環(huán)加成到環(huán)氧化物或氮雜環(huán)丙烷中,在溫和條件下具有優(yōu)異的區(qū)域選擇性。此外,由于CO2作為有效穩(wěn)定在MCM-41上的NHC的保護(hù)基團(tuán),使得催化劑可以通過簡單的過濾方法回收并重復(fù)使用多次而沒有明顯的活性損失[34]。

圖34
(四)熱反應(yīng)氮雜環(huán)丙烷環(huán)加成合成惡唑烷類化合物
2011年,Jakob Danielsson等人描述了甲亞胺葉立德與醛的1,3-偶極環(huán)加成反應(yīng)[35]。通過氮雜環(huán)丙烷的熱電環(huán)開環(huán)產(chǎn)生的偶氮甲堿葉立德與醛發(fā)生1,3-偶極環(huán)加成反應(yīng)得到了惡唑烷。隨后將惡唑烷水解成相應(yīng)的氨基醇,得到抗非對映異構(gòu)體作為主要產(chǎn)物。
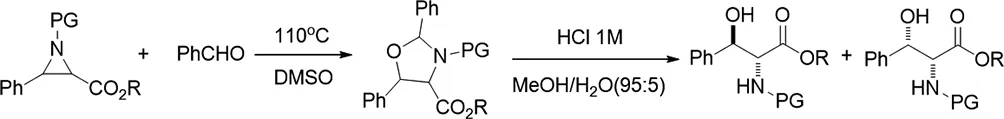
圖35
(五)熱反應(yīng)氮雜環(huán)丙烷環(huán)加成合成咪唑烷類化合物
2016年,Mohamed Ali Tabarki和Rafaa Besbes報(bào)道了通過多種N-取代的異硫氰酸酯用于合成咪唑烷-2-硫酮的環(huán)加成反應(yīng),涉及N-烷基氮雜環(huán)丙烷-2-羧酸酯的環(huán)擴(kuò)展[36]。后者通過完全區(qū)域和立體選擇性過程進(jìn)行開環(huán)和環(huán)化,得到目標(biāo)反式-咪唑烷-2-硫酮,這取決于N-取代基對氮雜環(huán)丙烷和異硫氰酸酯的空間和電子效應(yīng)。
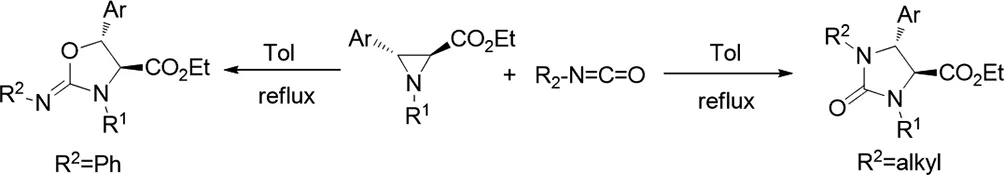
圖36
猜你喜歡
大自然探索(2023年7期)2023-11-14 13:08:06
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
智富時(shí)代(2018年3期)2018-06-11 16:10:44
浙江大學(xué)學(xué)報(bào)(工學(xué)版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
超硬材料工程(2016年1期)2016-02-28 22:20:04
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
合成化學(xué)(2015年4期)2016-01-17 09:01:27
應(yīng)用化工(2014年3期)2014-08-16 13:23:50
