枸橘藥材HPLC特征指紋圖譜研究
2020-11-12 12:57:12張亞雙王亞瓊張華鋒邢以文
中國合理用藥探索 2020年10期
關鍵詞:特征
付 妍,張亞雙,王亞瓊,張華鋒,陳 衛,周 堅,邢以文,薛 滿
(蘇州市藥品檢驗檢測研究中心,蘇州 215014)
枸橘為蕓香科(Rutaceae)植物枸橘PoncirustrifoliataL. Rafin.的近成熟果實,被《江蘇省中藥材標準(2016年版)》收載。有關研究表明[1-3],枸橘中含有黃酮類、揮發油類、香豆素類、生物堿類、有機酸類、萜類等化學成分。現代藥理學研究表明[4-13],枸橘具有抗腫瘤、抗過敏、抗病毒、調節胃腸道運動等作用。經對本草考證,枸橘又名綠衣枳實。有文獻報道測定枸橘中柚皮苷的含量[14],但若只測定其中一個組分的含量顯然不能全面有效地控制枸橘的質量。目前,特征指紋圖譜是控制中藥質量的有效方法之一。通過文獻檢索和研究發現,采用高效液相色譜法(high performance liquid chromatography,HPLC)的梯度洗脫程序能較好地分離枸橘中各組分,較全面地反映枸橘組成的整體信息,故選擇HPLC作為枸橘特征指紋圖譜的測定方法。
1 儀器與試藥
Waters高效液相色譜儀[沃特世科技(上海)有限公司,包括Waters-2489四元泵、Waters-2998二極管陣列檢測器、Empor化學工作站];KQ-250B型超聲清洗機(昆山超聲電子設備廠)。
甲醇為色譜純;水為重蒸餾水;冰醋酸為分析純;對照品柚皮苷(批號:110722-201111,純度:93.2%)和歐前胡素(批號:110722-201111,純度:99.6%)購于中國食品藥品檢定研究院;15批枸橘藥材由蘇州市天靈中藥飲片有限公司等6家企業提供,經蘇州市藥品檢驗檢測研究中心中藥室張華鋒主任鑒定為正品。
2 方法與結果
2.1 對照品溶液的制備
取柚皮苷對照品、歐前胡素對照品適量,精密稱定,加甲醇制成每1 ml各含柚皮苷150 μg、歐前胡素60 μg的溶液,即得。
2.2 供試品溶液的制備
取枸橘藥材粉末(過四號篩,批號:151219),約0.5 g,精密稱定,置具塞錐形瓶中,精密加入甲醇 50 ml,稱定重量,超聲處理(功率250 W,頻率50 kHz) 30 min,放冷,再稱定重量,加甲醇補足減失的重量,搖勻,濾過,取續濾液,即得。
2.3 色譜條件
Diamonsil Plus C18-A色譜柱(250 mm×4.6 mm,5 μm),以甲醇(A)-1%冰醋酸溶液(B)為流動相,按表1中的規定進行梯度洗脫,檢測波長283 nm,柱溫30 ℃。

表1 梯度洗脫表
2.4 色譜峰的確認
分別精密吸取對照品溶液與供試品溶液各10 μl,注入HPLC儀。供試品HPLC見圖1,圖中呈現6個特征峰,與參照物柚皮苷峰相對應的峰為峰2,計算各特征峰與峰2的相對保留時間,其相對保留時間應在規定值的±5%之內,其規定值為:0.97(峰1)、1.00(峰2)、1.22(峰3)、1.24(峰4)、1.62(峰5)、2.04(峰6)。

峰2(S):柚皮苷;峰5:歐前胡素圖1 枸橘藥材的HPLC圖
2.5 方法學驗證
2.5.1精密度試驗
取同一樣品,按“2.2”項下方法制備成供試液,進樣10 μl分析,一共進樣6次,考察各色譜峰相對保留時間和相對峰面積的一致性。結果:各特征峰的相對保留時間及相對峰面積的RSD均小于2.0%,說明該儀器的精密度良好。
2.5.2重復性試驗
取同一批號樣品(批號:15041403)共6份,按“2.2”項下方法制備成供試液,進樣10 μl,考察各色譜峰相對保留時間和相對峰面積的一致性。結果:各特征峰的相對保留時間及相對峰面積的RSD均小于4.0%,說明該方法的重復性良好。
2.5.3穩定性試驗
取同一批號樣品(批號:15041403),按“2.2”項下方法制備成供試液,分別于0、2、4、8和12 h進樣一次,共測定12 h,分別進樣10 μl,考察特征峰相對保留時間和相對峰面積的一致性。結果:各特征峰的相對保留時間及相對峰面積的RSD均小于3.0%,說明樣品供試液在12 h內穩定性良好。
2.5.4不同柱溫考察
采用Diamonsil Plus C18-A色譜柱(250 mm×4.6 mm,5 μm),分別考察了28、30和35 ℃。結果:不同柱溫對其分離度基本上沒有影響,考慮到色譜柱的耐受性以及分析時間的長短,故確定色譜柱的溫度為30 ℃。
2.5.5不同色譜柱考察
取同一批號樣品(批號:15041403),按“2.2”項下方法制備成供試液,采用Waters Symmetry Shield RP18色譜柱(250 mm×4.6 mm,5 μm)、Diamonsil Plus C18-A色譜柱(250 mm×4.6 mm,5 μm)、Boston Green ODS色譜柱(250 mm×4.6 mm,5 μm)進行測定。結果:Diamonsil Plus C18-A色譜柱(250 mm×4.6 mm,5 μm)檢出的色譜峰較多,且分離度良好,故選擇此色譜柱進行實驗。
2.6 特征指紋圖譜的建立
取15批枸橘藥材粉末,按“2.2”項下方法制備供試品溶液,按“2.3”項下方法檢測。將檢測結果導入軟件《中藥色譜指紋圖譜相似度評價系統》2004A版,15批枸橘藥材HPLC指紋圖譜見圖2,相對保留時間結果見表2。

圖2 15批枸橘藥材的HPLC特征指紋圖譜
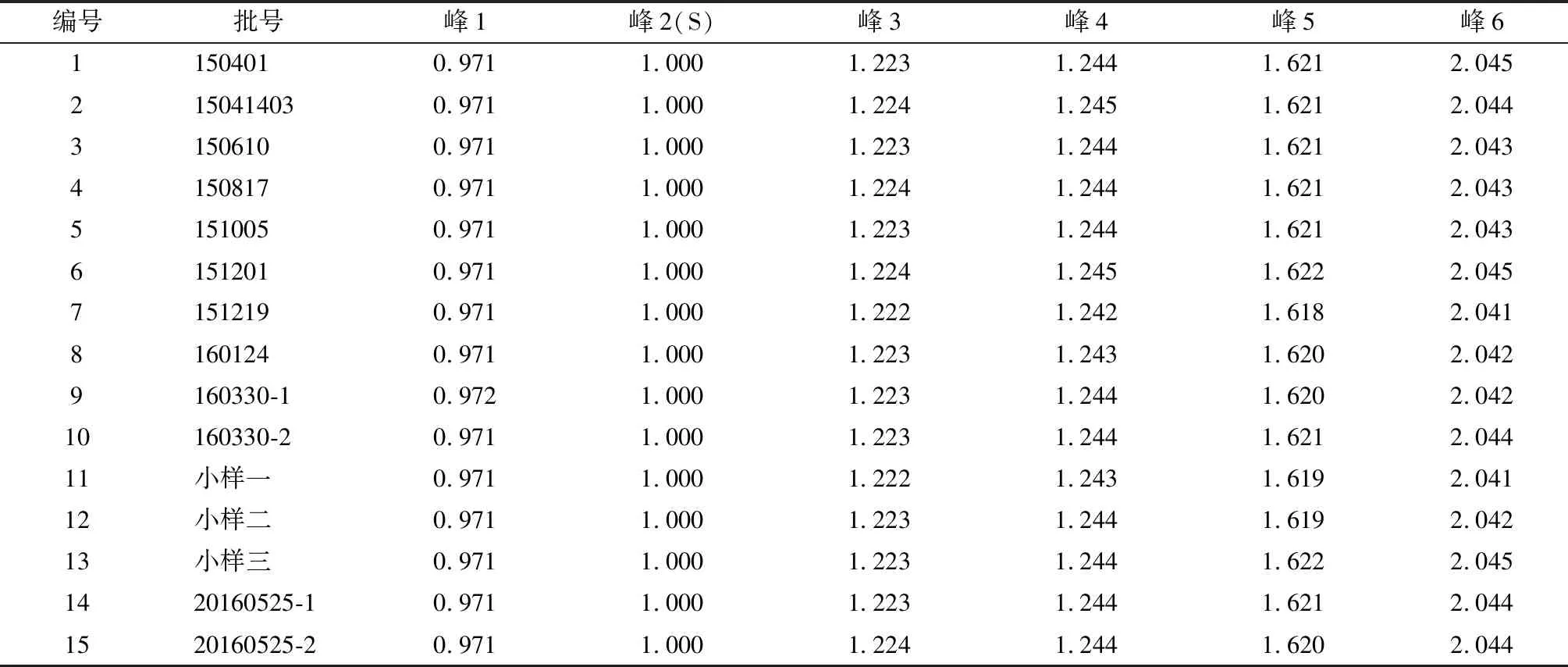
表2 15批樣品的相對保留時間min
3 討論
目前市場上枸橘藥材的質量良莠不齊,建立一個可靠的質量標準勢在必行。本研究建立的方法可以和枸橘藥材的含量測定一起作為控制枸橘藥材質量的標準,可以有效地監控枸橘藥材的組成與質量恒定性,為市場監管提供有效的技術支撐。
采用二極管陣列檢測器檢測,以峰數和分離度為指標,將樣品在200~400 nm進行掃描,分別考察了220、240、283、300和360 nm波長下的色譜峰,結果283 nm下的色譜峰分離較好,考慮到柚皮苷作為參考峰,且柚皮苷的最大吸收波長也在283 nm,因此最終確定283 nm作為檢測波長。
實驗中采用不同的提取溶劑,樣品的色譜峰基本一致,結合《江蘇省中藥材標準》2016年版枸橘項下,確定甲醇作為提取溶劑。樣品超聲處理不同時間,其HPLC圖基本一致,為提取完全,確定提取時間為30 min。
猜你喜歡
數學小靈通·3-4年級(2024年2期)2024-05-15 02:02:28
中學生數理化(高中版.高考數學)(2022年3期)2022-04-26 14:04:16
數學年刊A輯(中文版)(2020年1期)2020-05-19 00:30:36
空間科學學報(2020年2期)2020-04-01 03:50:40
瘋狂英語·新策略(2019年10期)2019-12-13 08:43:28
中等數學(2019年8期)2019-11-25 01:38:14
當代陜西(2019年10期)2019-06-03 10:12:04
新聞傳播(2018年11期)2018-08-29 08:15:24
數學小靈通·3-4年級(2017年9期)2017-10-13 08:10:54
廣西科技大學學報(2016年1期)2016-06-22 13:10:38
