鈣調控參與糖尿病小鼠冠狀動脈平滑肌收縮變化的機制
2020-11-18 07:39:44周夢園鄭丹琳秦曉玥李穗敏鄺素娟鄧春玉
中國藥理學通報 2020年11期
張 利,周夢園,鄭丹琳,秦曉玥,李穗敏,曾 鵬,鄺素娟,楊 慧,饒 芳,鄧春玉,3,4
[1. 華南理工大學生物科學與工程學院,廣東 廣州 510006; 2. 廣東省人民醫院(廣東省醫學科學院)醫學研究部臨床藥理重點實驗室,廣東 廣州 510080;3. 華南理工大學醫學院,廣東 廣州 510006;4. 廣東省心血管病研究所心血管內科,廣東 廣州 510100]
糖尿病患者首次確診時,多已伴有心血管并發癥,臨床上冠狀動脈粥樣硬化是其主要的致死原因[1]。糖尿病血管內皮功能障礙是其重要的病理生理特征,近年來大量研究表明,糖尿病的發生也伴隨著血管平滑肌細胞(vascular smooth muscle cells,VSMCs)功能異常,并影響著許多重要動脈的收縮。已有文獻報道,糖尿病大鼠主動脈和冠狀動脈的血管反應性發生變化,糖尿病主動脈的收縮反應減弱,冠狀動脈的舒張功能減弱,而收縮變化不明顯[2-3];然而,糖尿病小鼠胸主動脈血管收縮反應卻明顯增強[4],提示不同種屬和器官的糖尿病血管反應性變化存在差異,但糖尿病對小鼠冠狀動脈收縮影響的相關文獻報道甚少,有待進一步闡明。
胞內游離Ca2+在介導VSMCs收縮功能中起著重要作用[5]。VSMCs內Ca2+濃度變化受肌漿網內鈣釋放和胞外鈣內流的影響,其中L-型鈣通道和鈣庫操縱性鈣(store-operated calcium,SOC)通道是外鈣內流的主要途徑[6-7]。已有研究表明,糖尿病使L-型鈣通道和SOC通道功能發生改變[2-4],提示細胞內鈣調控異常與糖尿病血管反應性變化密切相關。因此,本研究通過給予不同的血管收縮劑5-羥色胺(5-hydroxyl tryptamine,5-HT)和血栓素A2類似物(9,11-dideoxy-11α, 9α-epoxymethanoprostaglandin,U46619)及不同信號通路阻斷劑硝苯地平(nifedipine)和毒胡蘿卜素(thapsigargin,TG),觀察糖尿病冠狀動脈血管張力變化,探討糖尿病對鈣調控參與冠狀動脈收縮反應性的差異,以研究糖尿病對小鼠冠狀動脈收縮的影響。
1 材料
1.1 實驗藥物U46619、5-HT(091M5163V)、硝苯地平(57H1139)、鏈脲佐菌素(streptozotocin,STZ;031M1287V)、毒胡蘿卜素(049M4032V)、咖啡因(Caffeine;71K1877)、EGTA(111K5411)均購于Sigma公司;其余試劑均為國產分析純。Krebs Henseleit(K-H)溶液(mmol·L-1):NaCl 119、NaHCO325、MgCl2·6H2O 1、KCl 4.7、KH2PO41.2、CaCl22.5、D-glucose 11.1;高鉀K-H溶液(mmol·L-1):KCl 60、NaCl 63.7、NaHCO325、MgCl2·6H2O 1、KH2PO41.2、CaCl22.5、D-glucose 11.1。Ca2+-free K-H溶液(mmol·L-1):NaCl 119、NaHCO325、MgCl2·6H2O 1、KCl 4.7、KH2PO41.2、D-glucose 11.1、EGTA 0.05。實驗所用溶液配好后均通混合氣(95% O2+5% CO2)充分飽和。
1.2 儀器620 M型小血管張力測定儀(丹麥DMT公司);PowerLab 8/30生物信號采集處理系統(澳大利亞AD公司);Stemi DV4 型體視顯微鏡(德國ZEISS公司);DK-8D 型電熱恒溫水槽(上海醫用恒溫設備廠);Advatage血糖儀和血糖試紙(Roche)。
1.3 實驗動物SPF級,8~9周齡C57BL/6小鼠,♂,共30只,購于江蘇集萃藥康生物科技有限公司,許可證號:SCXK(蘇)2018-0008,飼養于華南理工大學實驗動物中心。本研究所有動物實驗已通過廣東省人民醫院(廣東省醫學科學院)的動物實驗倫理審查(NoGDRE201208a)。
2 方法
2.1 糖尿病小鼠模型的建立將C57BL/6小鼠隨機分為模型組(model)和對照組(control),每組各15只。模型組和對照組通過腹腔注射分別注入等量STZ(50 mg·kg-1體質量)和溶劑(0.1mol·L-1檸檬酸-檸檬酸鈉緩沖液,pH 4.2~4.5),連續5 d給藥,同時給藥前禁食12 h不禁水,給藥后,再禁食2 h[4,8]。一周后使用羅氏血糖檢測試紙和血糖儀,通過尾靜脈采血法定期采集兩組小鼠的血糖水平變化,之后每隔1個月測量1次血糖水平,測量2次,以最后1次測量的血糖水平為準,模型組小鼠血糖高于16 mmol·L-1視為造模成功。
2.2 冠狀動脈環的制備血管環的制備同已發表文獻[9-10],簡而言之,通過CO2麻醉并處死小鼠后,快速取出心臟,放入4 ℃預冷的K-H(Krebs Henseleit)溶液中并借助體視顯微鏡去除冠狀動脈周圍的心肌組織,分離出冠狀動脈并剪成長度為1.8~2.0 mm血管環;用兩根直徑為40 μm的不銹鋼絲穿過血管環,并將其固定在張力測定儀浴槽內的兩個鉗夾上,同時鋼絲要保持平行。在37 ℃溫浴的K-H液中平衡30 min后,給予冠狀動脈1.2 mN的基礎張力,平衡60 min,期間每隔15 min換液1次,并使張力維持在1.2 mN。
2.3 藥物對冠狀動脈張力作用的檢測血管張力反應性的檢測同已發表文獻[9, 11]。對血管功能性檢測合格的血管,在浴液中加入單劑量5-HT(2 μmol·L-1),待血管收縮效應達到最大值后,用Ca2+-freeK-H液洗脫4次,給予1μmol·L-1硝苯地平和2 μmol·L-1TG孵育30 min,加入2.5 mmol·L-1CaCl2,觀察冠脈的血管反應性變化;用Ca2+-freeK-H液洗脫并給予1 μmol·L-1硝苯地平孵育,加入20 mmol·L-1Caffeine,觀察血管張力變化。
采用濃度梯度給藥的方法,在浴液中分別加入各濃度梯度的血管收縮劑5-HT (0.001~10 μmol·L-1)和U46619 (0.000 1~1 μmol·L-1),觀察血管收縮劑誘導血管張力的變化。當血管收縮效應達到最大值后,用K-H液洗脫4次,并加入1 μmol·L-1硝苯地平孵育30 min,重復上述5-HT和U46619濃度梯度給藥,觀察硝苯地平對血管收縮量效曲線的影響。當血管收縮效應達到最大值后,用Ca2+-free K-H液洗脫4次,并給予1 μmol·L-1硝苯地平孵育30 min,再次重復以上5-HT和U46619濃度梯度給藥,觀察在無鈣條件下硝苯地平對血管收縮張力變化的影響。
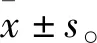
3 結果
3.1 糖尿病小鼠模型建立對照組與模型組小鼠分別注射等量溶劑和STZ,1周后,模型組小鼠出現明顯的“多飲、多食、多尿和體質量減輕”癥狀,對照組與模型組小鼠的體質量分別為(18.70±3.06)g和(29.68±2.59)g(n=12,P<0.01);與對照組(5.1±1.70)mmol·L-1相比,模型組小鼠的血糖水平(25.84±4.48)mmol·L-1明顯升高(n=12,P<0.01)。
3.2 小鼠冠狀動脈收縮的鈣調控機制高鉀刺激模型組冠狀動脈(2.84±0.40)mN(n=10)產生的收縮明顯低于對照組(1.86±0.53)mN(n=12)(P<0.01)。在Ca2+-free K-H液中給予L-型鈣通道阻斷劑硝苯地平和鈣泵阻斷劑TG孵育后,加入2.5 mmol·L-1CaCl2,未引起冠脈收縮;給予20 mmol·L-1Caffeine引起血管急劇收縮。提示冠脈的平滑肌收縮主要由經L-型鈣通道內流和肌漿網鈣釋放的鈣離子介導,SOC通道不參與冠脈收縮,見Fig 1。
3.3 糖尿病小鼠冠狀動脈對血管收縮劑5-HT和U46619的反應采用濃度梯度給藥法分別加入血管收縮劑5-HT(0.001~10 μmol·L-1)和U46619(0.000 1~1 μmol·L-1),發現5-HT與U46619均可誘導小鼠冠狀動脈呈明顯的濃度依賴性收縮,模型組小鼠冠狀動脈平滑肌收縮反應明顯低于對照組。5-HT誘導模型組冠狀動脈量效曲線的Emax以及pEC50(Emax:64.12±18.23,n=9;pEC50:6.42±0.25,n=9)均明顯低于對照組(Emax:112.75±9.33,n=10;pEC50:7.12±0.20,n=10)(P<0.01)。U46619誘導模型組冠狀動脈量效曲線的pEC50(7.17±0.34,n=9)明顯低于對照組(7.78±0.33,n=8)(P<0.01),而Emax在兩組間差異無顯著性(P>0.05),見Fig 2。
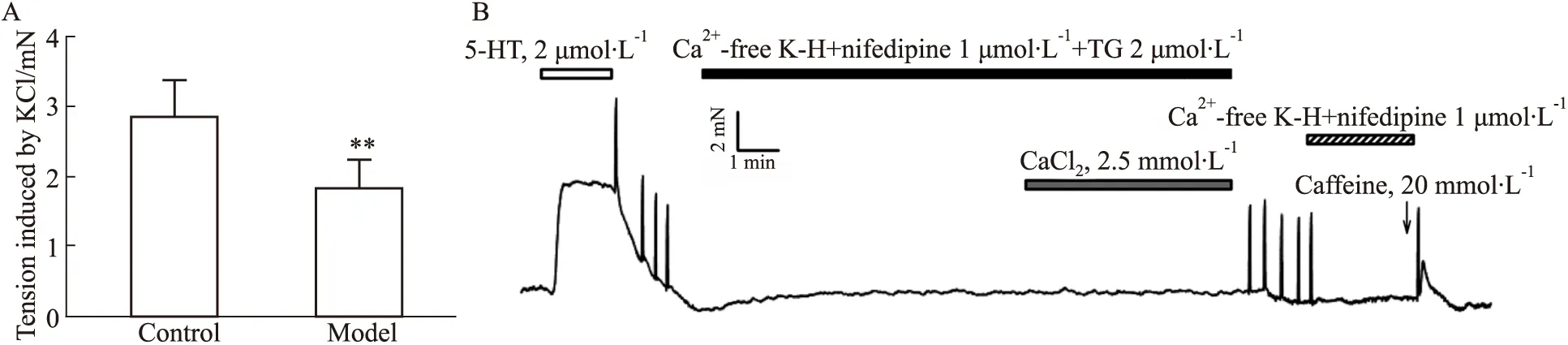
Fig 1 The mechanism of calcium handling involved in constriction of coronary artery in mice A: High potassium-evoked contraction was stronger in the coronary arterial rings of wild-type mice than of diabetic mice (n control=12, nmodel=10); B: Coronary arteries were washed in Ca2+-free buffer in the presence of nifedipine (1 μmol·L-1) and TG (2 μmol·L-1) for 30 min, and there was no response to CaCl2 at 2.5 mmol·L-1 in coronary arteries of mice, and caffeine at 20 mmol·L-1 significantly evoked constriction of coronary arteries in mice(n=3). **P<0.01 vs control.

Fig 2 The concentration-dependent vasoconstriction induced by 5-HT and U46619 in coronary arterial rings of diabetic mice Representative recording of 5-HT(A)- and U46619 (C)-elicited concentration-dependent contraction in the coronary arterial rings of wild-type and diabetic mice. The concentration-dependent vasoconstriction induced by 5-HT (B) and U46619 (D) in the coronary arterial rings of wild-type and diabetic mice. 5-HT: ncontrol=10, nmodel=8; U46619: ncontrol=10, nmodel=9. **P<0.01 vs control.
3.4 硝苯地平對血管收縮劑誘導糖尿病小鼠冠狀動脈收縮反應的影響加入硝苯地平(1 μmol·L-1)孵育后,采用濃度梯度給藥法分別給予血管收縮劑5-HT(0.001~10 μmol·L-1)和U46619(0.000 1~1 μmol·L-1),記錄冠狀動脈張力變化。結果顯示,硝苯地平可明顯抑制血管收縮劑誘導冠狀動脈的收縮反應;模型組冠狀動脈收縮反應明顯低于對照組,硝苯地平對模型組冠狀動脈收縮反應的抑制幅度明顯降低(P<0.01)。5-HT誘導模型組冠狀動脈的量效曲線的Emax(6.41±6.73,n=9)明顯低于對照組(15.75±4.15,n=10)(P<0.05),而兩組間的pEC50差異無顯著性(P>0.05)。在U46619誘導冠狀動脈收縮反應,兩組的Emax和pEC50差異均無統計學意義(P>0.05),見Fig 3。
3.5 肌漿網Ca2+釋放在糖尿病小鼠冠狀動脈收縮反應中的變化在Ca2+-free K-H溶液中加入硝苯地平(1 μmol·L-1)孵育后,采用濃度梯度給藥法分別給予5-HT(0.001~10 μmol·L-1)和U46619(0.000 1~1 μmol·L-1),記錄冠狀動脈張力變化。結果顯示,在細胞外液無Ca2+條件下,模型組小鼠冠狀動脈的收縮量效曲線明顯右移。在5-HT和U46619誘導冠狀動脈收縮反應中,兩組間的Emax和pEC50均無明顯差異(P>0.05),見Fig 4。
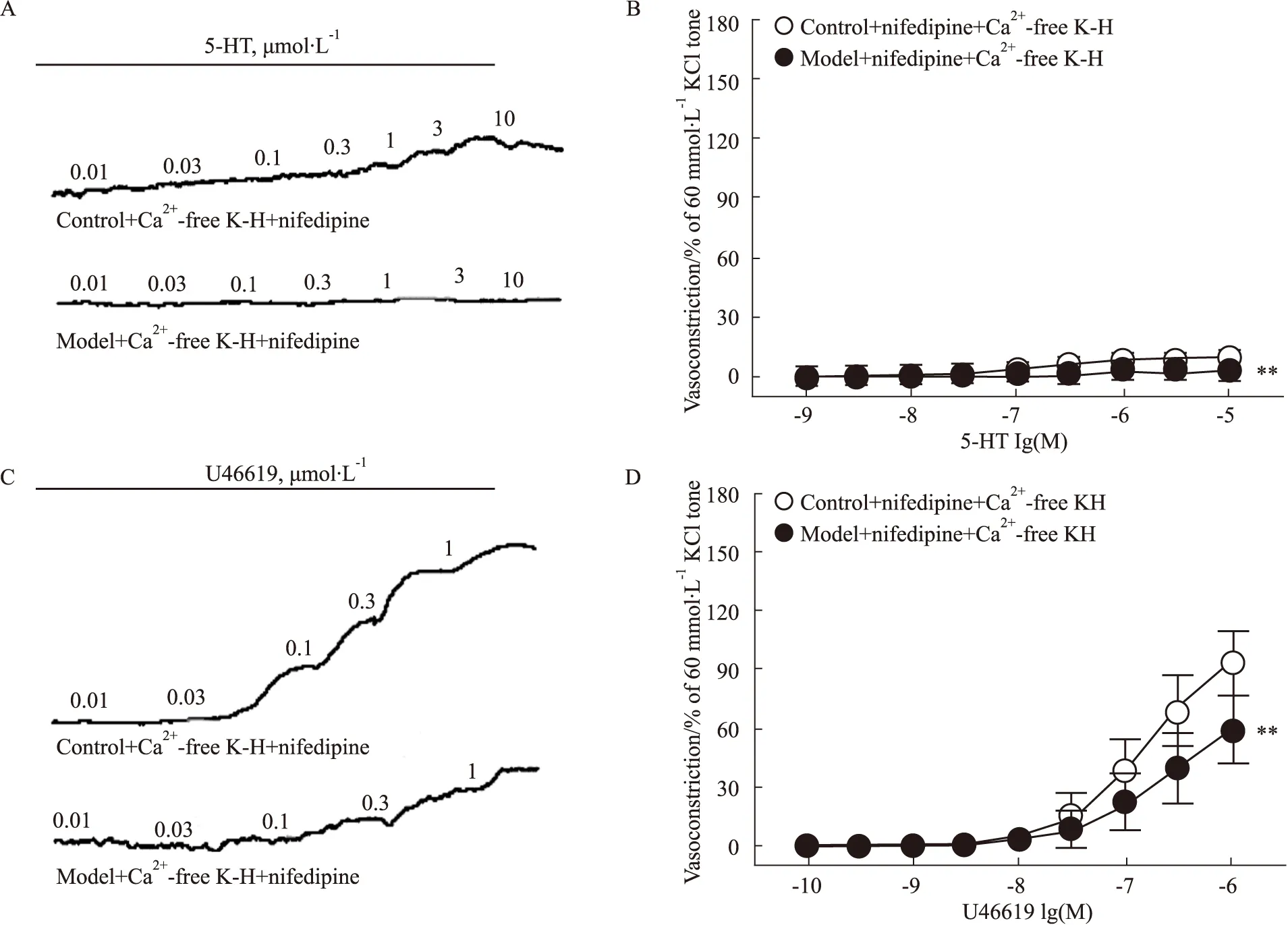
Fig 4 Effect of sarcoplasmic reticulum calcium on concentration-dependent vasoconstriction induced by 5-HT and U46619 in coronary arterial rings of diabetic mice Representative recording of 5-HT(A)- and U46619 (C)- elicited concentration-dependent contraction in the coronary arterial rings of wild-type and diabetic mice. In Ca2+-free buffer, effect of nifedipine (1 μmol·L-1) on concentration-dependent vasoconstriction induced by 5-HT (B) and U46619 (D) in the coronary arterial rings of wild-type and diabetic mice. 5-HT: ncontrol=8, nmodel=8; U46619: ncontrol=7, nmodel=9. **P<0.01 vs control.
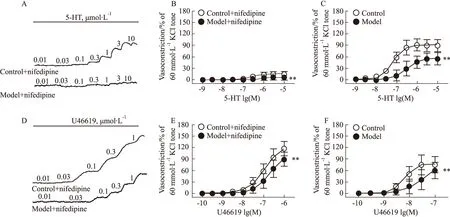
Fig 3 Effect of nifedipine on concentration-dependent vasoconstriction induced by 5-HT and U46619 in coronary arterial rings of diabetic mice Representative recording of 5-HT(A)- and U46619 (D)- elicited concentration-dependent contraction in the coronary arterial rings of wild-type and diabetic mice. The effect of nifedipine (1 μmol·L-1) on concentration-dependent vasoconstriction evoked by 5-HT (B) and U46619 (E) in the coronary arterial rings of wild-type and diabetic mice. The concentration-dependent vasoconstriction induced by 5-HT (C) and U46619 (F) inhibited by nifedipine (1 μmol·L-1) in the coronary arterial rings of wild-type and diabetic mice. 5-HT: ncontrol=11, nmodel=9; U46619: ncontrol=10, nmodel=9. **P<0.01 vs control.
4 討論
VSMCs的收縮依賴于細胞內Ca2+濃度(intracellular calcium concentration,[Ca2+]i)的增加,[Ca2+]i的增加主要通過胞外鈣內流和肌漿網內鈣釋放介導[5]。細胞外Ca2+內流途徑主要包括與膜電位有關的L-型鈣通道以及與鈣庫耗竭有關的SOC通道[6-7]。本課題組已研究報道2型糖尿病大鼠主動脈血管收縮反應性減弱與L型鈣通道下調有關,SOC通道介導的收縮明顯增強[2];但SOC通道不參與大鼠冠狀動脈的收縮[12]。因此本實驗通過不同血管收縮劑以及信號通路阻斷劑,主要研究在有或無L-型鈣通道介導下糖尿病小鼠冠脈收縮的差異性以及SOC通道和肌漿網鈣釋放功能的變化。
血管收縮劑5-HT和血栓素A2類似物U46619分別作用于VSMCs膜上的5-HT2A受體和血栓烷素(TP)受體[13-14],通過Gq蛋白激活磷脂酶C(PLC),促使磷脂酰肌醇二磷酸(PIP2)水解,生成三磷酸肌醇(IP3)和二酰基甘油(DAG),IP3與肌漿網上的IP3受體結合觸發肌漿網內Ca2+釋放,使膜去極化二次激活L-型鈣通道,同時肌漿網內Ca2+耗竭可激活SOC通道,使細胞外鈣內流,引起血管收縮;DAG可以通過作用于非選擇性鈣離子通道參與VSMCs收縮,另一方面還可以作用于蛋白激酶C(PKC),增加鈣敏感性促進血管收縮[10, 15]。
為了闡明小鼠冠狀動脈收縮的鈣調控機制,采用高鉀刺激血管引起收縮,提示L-型鈣通道參與冠脈收縮;在細胞外液無Ca2+條件下加入硝苯地平阻斷L-型鈣通道介導的血管收縮以及肌漿網鈣泵抑制劑TG進行孵育,發現加入CaCl2未引起冠脈收縮,而此時介導外鈣內流參與血管收縮的途徑為SOC通道,提示SOC通道不參與小鼠冠脈的收縮。同時實驗中發現給予Caffeine后冠脈急劇收縮,而Caffeine通過誘導肌漿網Ca2+釋放增加[Ca2+]i引起血管收縮,提示肌漿網鈣釋放參與小鼠冠脈收縮。
研究表明,糖尿病對血管平滑肌細胞的收縮功能產生影響[2-4],為了闡明糖尿病對小鼠冠狀動脈血管張力的影響,本實驗利用血管收縮劑5-HT和U46619誘導冠脈收縮,發現糖尿病小鼠冠狀動脈收縮反應性較對照組明顯減弱,提示糖尿病影響冠狀動脈的收縮。實驗中發現高鉀刺激糖尿病小鼠冠狀動脈引起的收縮反應明顯減弱,而高鉀使VSMCs膜去極化,使與膜電位有關的L-型鈣通道開放,細胞外Ca2+流入胞內,引起血管收縮,提示糖尿病影響L-型鈣通道介導的血管收縮。為了進一步驗證糖尿病狀態下鈣調控機制的相關變化,采用L-型鈣通道阻斷劑硝苯地平進行孵育,加入收縮劑誘導血管收縮,發現硝苯地平對糖尿病組血管收縮反應的抑制幅度明顯低于對照組,進一步說明糖尿病小鼠冠狀動脈L型鈣通道調控功能下調;并且糖尿病組對激動劑誘導的血管收縮明顯低于對照組,提示糖尿病影響非L-型鈣通道介導的血管收縮。隨后,我們探討糖尿病狀態下肌漿網鈣釋放功能的變化,發現在含硝苯地平的無鈣K-H溶液中,收縮劑誘導糖尿病組的血管反應性明顯低于對照組,提示糖尿病小鼠肌漿網鈣釋放功能下調。
綜上所述,本研究發現參與小鼠冠狀動脈平滑肌收縮的鈣調控機制主要包括L-型鈣通道和肌漿網鈣釋放,SOC通道不參與其收縮;糖尿病小鼠冠狀動脈對血管收縮劑的反應性明顯減弱,與L-型鈣通道和肌漿網鈣釋放功能下調有關。此外,Yokota等[16-17]研究表明,糖尿病患者對5-HT和血管緊張素II(Ang II)誘導血管收縮反應性增強與VSMCs膜上5-HT2A受體和AT1受體表達上調有關,糖尿病小鼠冠脈張力的變化是否也與VSMCs膜上相應受體的表達有關,本實驗中未涉及,有待進一步研究。
猜你喜歡
興趣閱讀·興趣作文與閱讀(低年級)(2025年8期)2025-08-18 00:00:00
中老年保健(2022年5期)2022-08-24 02:35:42
中老年保健(2022年1期)2022-08-17 06:14:56
中老年保健(2021年5期)2021-08-24 07:07:20
中老年保健(2021年9期)2021-08-24 03:51:04
中老年保健(2021年7期)2021-08-22 07:42:16
中老年保健(2021年11期)2021-08-22 03:15:16
學苑創造·A版(2020年9期)2020-10-13 09:41:02
小學生學習指導(低年級)(2017年10期)2017-10-10 01:00:05
云南中醫學院學報(2014年3期)2014-07-31 18:57:34
