三棱飲片薄層色譜鑒別及其總黃酮的含量測定
2020-11-19 01:06:28蔣立勇鐘方曉李克明張會敏
中國民族民間醫藥 2020年19期
關鍵詞:黃酮
蔣立勇 董 學 鐘方曉 李克明 張會敏
1.山東省泰安市新泰市中醫醫院,山東 新泰 271200;2.山東省中醫藥研究院,山東 濟南 250014
三棱為黑三棱科黑三棱屬植物黑三棱Sparganiumstoloniferum(Graebn.)Buch.的干燥塊莖[1],主要分布于我國東北、黃河、長江中下游流域,野生資源較為豐富。其味苦、性平,入肝、脾經,具有破血行氣、消積止痛等功效,用于癥瘕痞塊、瘀血經閉、食積脹痛。三棱中含有的化學成分有黃酮類、揮發油類、皂苷類[2-4]等。現代藥理研究表明三棱總黃酮具有較強的抗血小板聚集[5]、抗血栓和鎮痛[6]作用并且具有抑制腫瘤細胞A549、MCF-7、宮頸癌Hela細胞的細胞增殖活性等作用[7]。目前,2015版《中國藥典》僅僅規定了三棱飲片的性狀,對薄層鑒別、水分和浸出物進行了考察,沒有進行含量測定研究。在國內外研究報道中,張建中等[8-9]對三棱飲片的薄層色譜和總黃酮含量也展開了一定的研究,但研究內容還不夠完善,薄層展開方法以及結論還不夠詳實。因此,本研究在此基礎上運用系列薄層色譜法對16批不同產地的三棱飲片進行研究,優化三棱飲片的薄層鑒別方法,完善和提高三棱飲片薄層鑒別的準確性與可靠性,同時運用紫外可見分光光度法分別對三棱飲片中的總黃酮含量進行研究,建立三棱飲片總黃酮的含量測定方法。
1 儀器與材料
1.1 實驗儀器 939薄層制板器(重慶市南岸貝爾德儀器技術廠), ZX-002紫外分光光度計(日本島津),FW177型粉碎機(天津市泰斯特儀器有限公司)
1.2 實驗材料 蘆丁對照品(中國食品藥品生物制品檢定研究院,批號100080-200306),三棱對照藥材(中國食品藥品生物制品檢定研究院,批號121521-201504),乙醇(分析純,天津市富宇精細化工有限公司),亞硝酸鈉(分析純,天津市博迪化工有限公司),硝酸鋁(分析純,天津市博迪化工有限公司),氫氧化鈉(分析純,國營山東單縣有機化工廠)。
三棱飲片來自于各大藥店和藥材市場。經鐘方曉研究員鑒定為黑三棱科黑三棱屬植物黑三棱Sparganiumstoloniferum(Graebn.)Buch.的干燥塊莖,其飲片如圖1所示,具體來源見表1。

圖1 三棱飲片圖

表1 三棱飲片來源
2 方法與結果
2.1 薄層色譜鑒別
2.1.1 供試品溶液及對照藥材溶液的制備 取本品粉末(過60目篩)1.0 g,加甲醇3 mL,浸泡過夜,取上清液作為供試品溶液。另取三棱對照藥材1.0 g,同法制備對照藥材溶液。
2.1.2 藥典薄層鑒別方法 吸取供試品溶液及對照藥材溶液各5 μL,以樣品1~8、9(三棱對照藥材)、10~17為點樣順序,分別點于同一以羧甲基纖維素鈉為黏合劑的硅膠G薄層板上,以石油醚:乙酸乙酯(v∶v=4∶1, 60~90 ℃)為展開劑,飽和30 min,展開,取出,晾干。置紫外光燈下(365 nm)檢視,如圖2所示。
由圖2可知,15個樣品溶液(4號樣品溶液除外)在與對照藥材溶液相同位置均有一組相同的藍色熒光斑點1(Rf值0.33),這些斑點強弱差異較大,1、2、6、7、10、12~17斑點較弱,4號樣品未發現該熒光斑點,按照藥典的展開條件這些樣品可能鑒別為劣藥甚至假藥,為了提高鑒別的準確度,有必要對三棱飲片進行系列薄層色譜研究,來尋找更優的三棱飲片薄層鑒別方法。
2.2.3 優化后的薄層色譜方法 吸取供試品溶液及對照藥材溶液各5 μL,以1~8、9(三棱對照藥材)、10~17為點樣順序,分別點于同一以羧甲基纖維素鈉為粘合劑的硅膠G薄層板上,以石油醚:乙酸乙酯:甲酸(v∶v∶v=3∶1∶0.1,60~90 ℃)為展開劑,飽和30 min,展開,取出,晾干。置紫外光燈下(365 nm)下檢視,如圖3所示。再噴以5%硫酸乙醇,熱風吹至斑點清晰,在可見光下觀察,如圖4所示。再次置紫外光燈下(365 nm)檢視,如圖5所示。
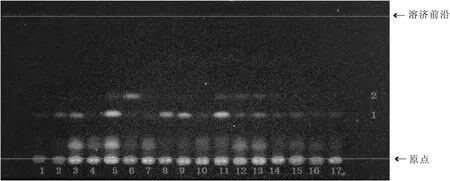
1~8.三棱樣品;9.三棱對照藥材;10~17.三棱樣品圖2 2015年版中國藥典方法三棱薄層色譜圖
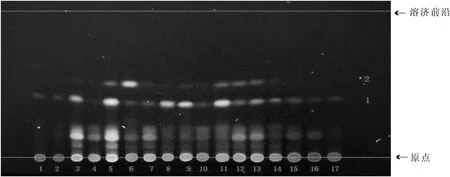
1~8.三棱樣品;9.三棱對照藥材;10~17.三棱樣品圖3 新建方法三棱薄層色譜圖
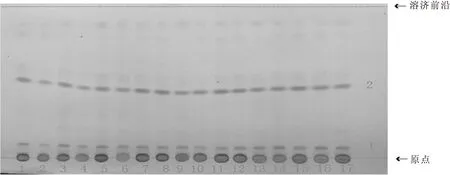
1~8.三棱樣品;9.三棱對照藥材;10~17.三棱樣品圖4 可見光下5%硫酸乙醇加熱顯色后三棱薄層色譜圖
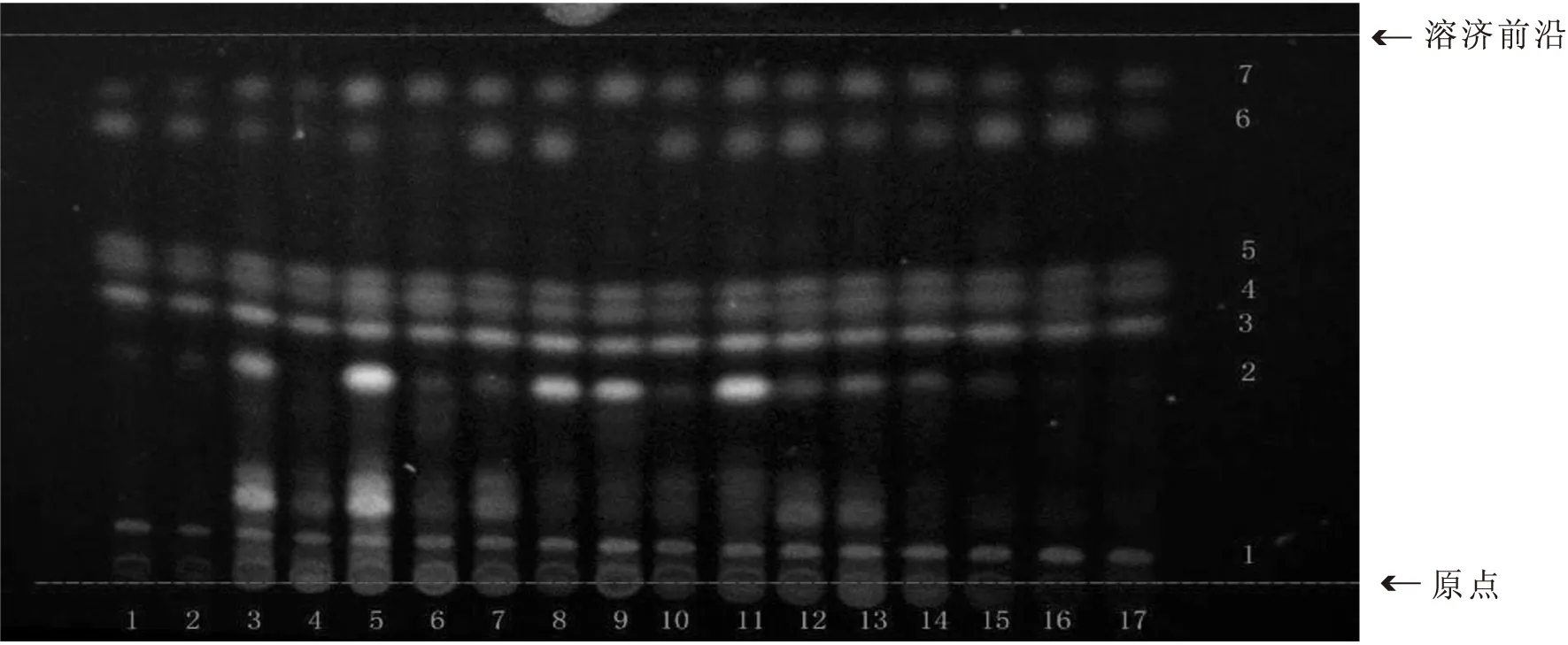
1~8.三棱樣品;9.三棱對照藥材;10~17.三棱樣品圖5 紫外光下5%硫酸乙醇加熱顯色后三棱薄層色譜圖
由圖3可知,在石油醚:乙酸乙酯:甲酸(v∶v∶v=3∶1∶0.1,60~90 ℃)展開條件下,紫外燈下直接觀察,除4號樣品溶液外,其余15個樣品溶液在與對照藥材溶液相同位置均有相同的藍色熒光斑點,但斑點顏色強弱差異較大;由圖4可知,在可見光下,16個樣品溶液與對照藥材溶液在相同位置有兩個相同的紅色斑點1(Rf值0.05)和2(Rf值0.46),且斑點強度相近;由圖4可知,1(Rf值0.05)、3(Rf值0.46)、4(Rf值0.50)、5(Rf值0.52)、7號斑點(Rf值0.90)為16個樣品溶液和對照藥材溶液的共有斑點,且熒光強度接近;2號斑點(Rf值0.38)為藥典條件下所觀察的熒光斑點,樣品溶液與對照藥材溶液熒光斑點顏色強弱差異較大;6號斑點(Rf值0.85)為15個樣品溶液的共有斑點(4號樣品溶液除外),但對照藥材溶液沒有。
由上述研究可見,按照藥典薄層方法得到的信息量較少,無法準確的鑒別出三棱飲片的質量真偽優劣,可能會得出錯誤的結論,而采用改良后的薄層展開方法后,至少存在5個以上共有斑點,可以通過這些共有斑點快速準確的綜合評價三棱飲片的真偽優劣。
2.2 三棱飲片總黃酮的含量測定[10]
2.2.1 對照品溶液的制備 精密稱取在120 ℃干燥至恒重的蘆丁標準品5.22 mg,置 50 mL容量瓶中,加60%乙醇適量,采用超聲處理,使其溶解,放冷,加60%的乙醇至刻度,搖勻,即得(每1 mL中含蘆丁0.1044 mg)。
2.2.2 測定波長的確定 精密量取6 mL蘆丁對照品溶液,置于25 mL容量瓶中,加水6 mL,加5%亞硝酸鈉溶液1.0 mL,搖勻,放置6 min;加入10%硝酸鋁溶液1.0 mL,搖勻,放置6 min;加入4%氫氧化鈉試液5 mL,再加至刻度,搖勻,放置15 min。以不加蘆丁對照品同上法平行操作的相應溶液為空白對照,進行240~600 nm全波段掃描。結果顯示,在500 nm 處有最大吸收波長,故選擇波長500 nm 處進行含量測定。
2.2.3 標準曲線的制備 精密量取蘆丁對照品溶液2 、4 、6 、8 、10 、12 mL,分別置25 mL量瓶中,后按2.2.2的方法制備樣品,以不加蘆丁對照品同上法平行操作的相應溶液為空白對照,在最大吸收波長處測定吸光度。以吸光度為縱坐標,濃度為橫坐標,繪制標準曲線,再用最小二乘法進行線性回歸,以吸光值對濃度進行線性回歸分析, 求得回歸方程為A=0.1062C-0.0053(R2=0.9995),表明蘆丁對照品溶液濃度在0.008~0.050 mg·mL-1范圍內與吸光度呈良好的線性關系。
2.2.4 提取條件的選擇
2.2.4.1 乙醇濃度的選擇 精密稱取三棱粉末(5號樣品)1.0 g,平行稱定3份,分別置于100 mL錐形瓶中,精密加入25 mL不同濃度的乙醇(第1份40%乙醇,第2份60%乙醇,第3份80%乙醇),后按2.2.2的方法制備樣品,在最大吸收波長處測定吸光度。 由表2可見,用60%乙醇提取后總黃酮含量最高,故選用60%乙醇溶液提取。
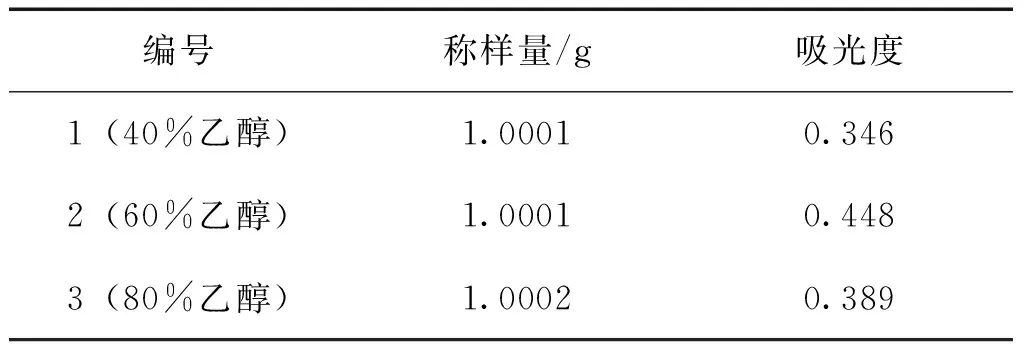
表2 不同濃度乙醇的吸光度值
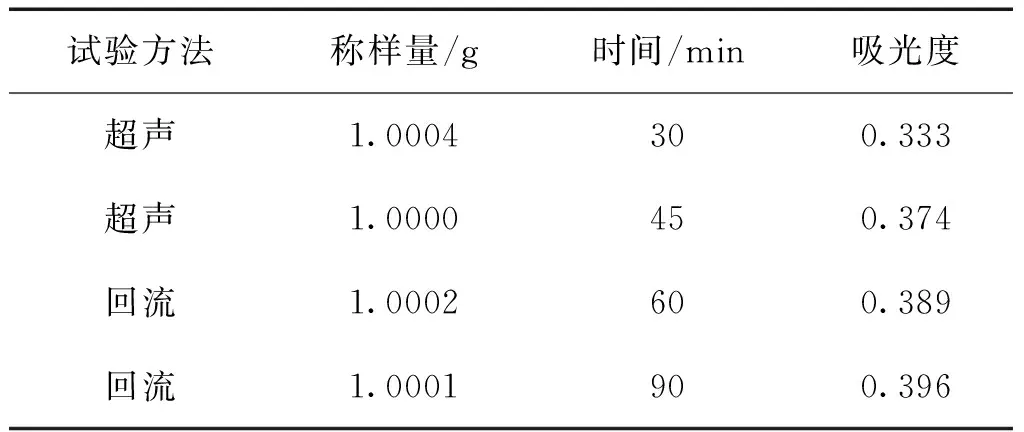
表3 不同提取時間及提取方法的吸光度
2.2.4.2 提取時間及提取方法的選擇 精密稱取三棱粉末(5號樣品)1.0g,平行稱定4份,分別置于100 mL錐形瓶中,精密加入25 mL的60%乙醇,稱重,其中兩份樣品溶液超聲提取,另兩份加熱回流提取,測定吸光度。由表3可見,回流提取的總黃酮含量高于超聲提取的,但回流1 h與回流1.5 h差距不大,故選加熱回流1h為提取方法。結果表明,以60%乙醇溶液,加熱回流提取1 h為最佳提取工藝。
2.2.5 供試品溶液的制備 精密稱取三棱粉末(3號樣品)1.0 g,置于100 mL錐形瓶中,精密量取60%乙醇25 mL,稱重,加熱回流提取1h,放冷后,用60%乙醇溶液補足損失的重量,過濾,濾液為供試品溶液。
2.2.6 精密度試驗 精密量取蘆丁對照品溶液6 mL,置25 mL量瓶中,按照2.2.3的方法,自“各加水6 mL”起依法測定吸光度,連續測6次,按吸光度計算標準偏差(SD)為0.000471,相對標準偏差(RSD)為0.173%。
2.2.7 穩定性試驗 精密量取供試品溶液5 mL,置25 mL量瓶中,按照2.2.3的方法,自“各加水6 mL”起依法測定吸光度,分別測定顯色后放置10、20、30、40、50、60 min 供試品溶液的吸光度,按吸光度算得其在1 h內標準偏差(SD)為0.005,相對標準偏差(RSD)為3.00%。
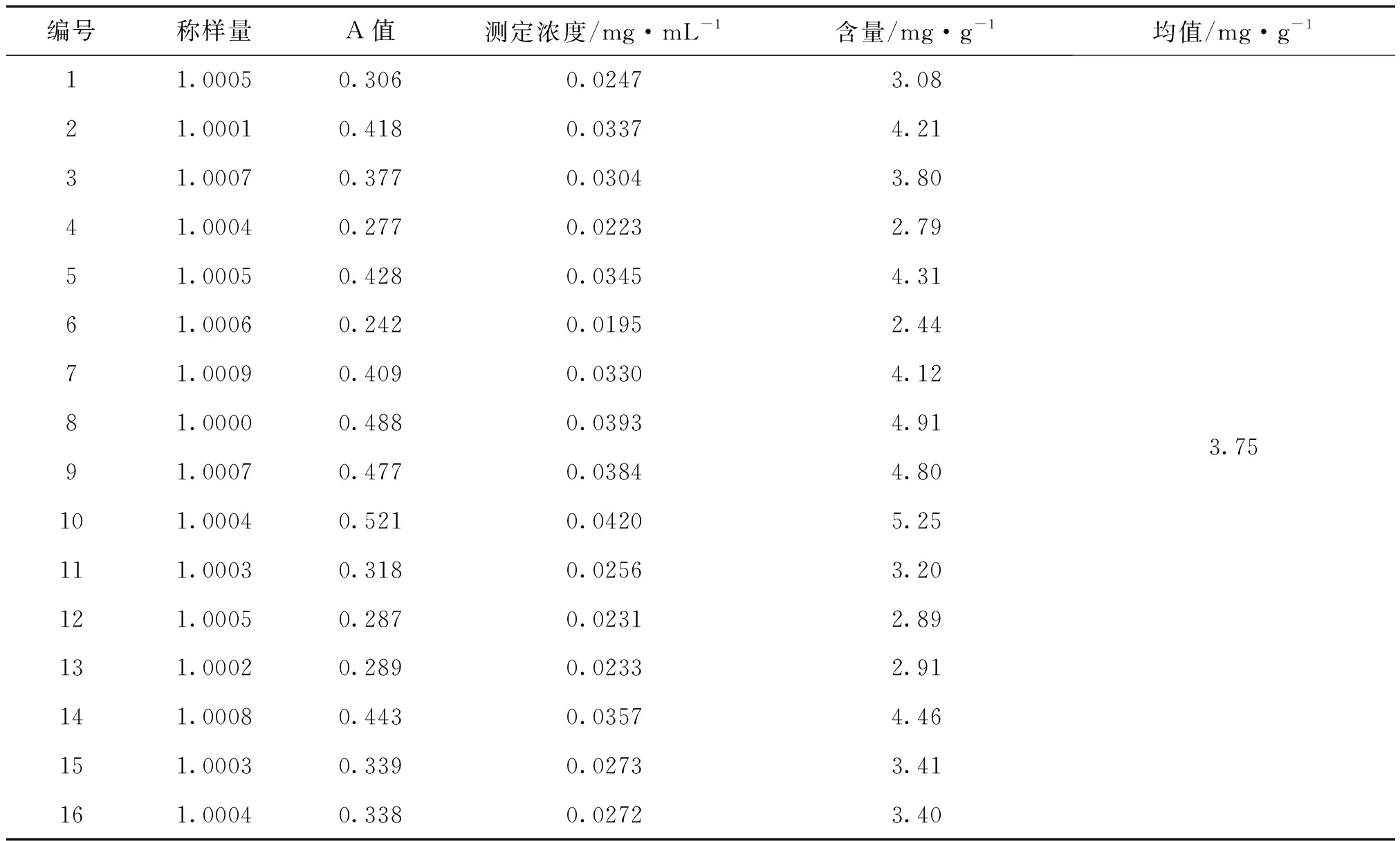
表4 不同來源的三棱飲片中總黃酮的含量
2.2.8 重復性試驗 精密稱取6份同一袋樣品各1.0 g,按照2.2.5的方法平行操作制備6份供試品溶液,分別精密吸取6份供試品溶液各5 ml,置25 mL量瓶中,按照2.2.3的方法,自“各加水6 mL”起依法測定吸光度,按含量計算相對標準偏差(RSD)為2.49%。
2.2.9 加樣回收率試驗 稱取已知含量的樣品約0.50 g,精密稱定,平行6份,分別加入一定量的蘆丁對照品,按2.2.5制得供試品溶液。精密量取供試品溶液5 mL,置于25 mL錐形瓶中,按照2.2.3的方法,自“各加水6 mL”起依法測定吸光度。用實值除以理論值計算回收率,再計算平均回收率與相對標準偏差(RSD)。算得平均回收率為98.9%,RSD為1.49%。表明該含量測定方法的誤差以及操作過程的損失較小,可靠性較高。
2.2.10 三棱中總黃酮的含量測定 精密稱取本品粉末1.0 g,置100 mL錐形瓶中,精密加入60%乙醇25 mL,浸泡,稱重,加熱回流提取1 h,放冷,60%乙醇補足損失重量,搖勻,濾過,濾液為供試品溶液。精密量取供試品溶液5 mL,置25 mL容量瓶中,按照2.2.3的方法,測定吸光度,計算供試品中總黃酮的含量(mg·g-1),再按含量計算均值,結果表4。
16批三棱藥材總黃酮的平均含量為3.75 mg·g-1。最低含量為2.43 mg·g-1,最高含量為5.25 mg·g-1。
3 結論
通過實驗可知,在石油醚-乙酸乙酯-甲酸(3∶1∶0.1)的展開條件下,可顯現5組與對照藥材溶液相同的共有斑點,且這些斑點更為清晰,更具有代表性,可用于三棱飲片的快速鑒別。在2015版《中國藥典》的薄層展開條件下,三棱飲片在紫外光下僅能觀察到一組熒光斑點,而本實驗建立的方法,在紫外光下5%硫酸乙醇加熱顯色后三棱薄層色譜圖顯示7組熒光斑點,斑點多且清晰,專屬性強。運用紫外可見分光光度法可以快速測定三棱飲片中總黃酮的含量。
猜你喜歡
四川蠶業(2021年2期)2021-03-09 03:15:32
四川蠶業(2021年3期)2021-02-12 02:38:46
中成藥(2018年11期)2018-11-24 02:57:00
中成藥(2017年8期)2017-11-22 03:19:40
中成藥(2017年10期)2017-11-16 00:50:13
中成藥(2017年4期)2017-05-17 06:09:50
哈爾濱醫藥(2016年1期)2017-01-15 13:43:16
天然產物研究與開發(2016年11期)2016-06-15 20:29:17
湖南師范大學自然科學學報(2015年1期)2015-02-27 14:50:04
安徽醫藥(2014年12期)2014-03-20 13:15:15
