基于PKD1/HDAC7軸研究丹參酚酸B對大鼠缺血心肌組織的保護作用
2020-12-17 12:05:14劉暖,楊雷,劉萍
中國藥理學通報 2020年12期
劉 暖,楊 雷,劉 萍
(南陽理工學院河南省張仲景方藥與免疫調節重點實驗室,河南 南陽 473004)
心肌梗死(myocardial infarction,MI)等缺血性疾病會引發大量心肌細胞死亡,導致患者左心室不良重構、心功能受損、生活質量低下[1]。丹參為治療MI等缺血性心肌疾病的經典中藥材之一,丹參酚酸B(salvianolic acid B,SAB)屬于水溶性酚酸類化合物,為丹參的代表性單體成分[2]。課題組前期的實驗研究證實,蛋白激酶D1(protein kinase D1,PKD1)具有促進血管新生的作用[3-4],并且可以下調MI后心肌膠原蛋白的表達,進而逆轉MI后的心室重塑,保護缺血受損的心肌[5]。PKD1在體內發揮上述對缺血心肌的部分保護效應需要依賴和IIa類組蛋白去乙酰化酶7(group IIa histone deacetylase 7,HDAC7)的結合,形成PKD1/HDAC7軸才能夠完成[6]。CID755673為PKD1的特異性阻斷劑。本研究擬分析MI大鼠SAB給藥及應用阻斷劑14 d后心肌組織的形態學變化及PKD1和HDAC7蛋白的表達,闡述SAB是否通過調控PKD1/HDAC7軸保護缺血受損的心肌組織。
1 材料
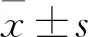
1.1 實驗動物40只Wistar大鼠,SPF級,♂,8周齡,體質量(200±20)g,購自北京維通利華公司,動物生產許可證號:SCXK(京)2016-0011;動物質量合格證號:11400700315978。
1.2 藥物與試劑SAB(上海陶素公司,批號:115939-25-8),CID755673(簡稱CID,美國MCE公司,批號:2018276);TUNEL染色試劑盒(上海翊圣公司,批號:40308ES20);PKD1(上海圣克魯斯公司,批號:Sc-1796)、HDAC7(武漢三鷹生物公司,批號:DF6435)的一抗抗體;Cy3熒光標記的羊抗兔IgG二抗(武漢博士德公司,批號:BA1032);其它試劑為國產分析純。
1.3 主要儀器LSM 800激光共聚焦顯微鏡和ZEN 2.3 lite分析軟件(德國蔡司公司);CUT6062病理切片機和MTMI全密閉自動脫水機(德國賽利公司)。
2 方法
2.1 動物造模大鼠隨機分為假手術組(Sham)、心梗模型組(MI)、SAB給藥組(SAB)和CID阻斷劑組(CID)。結扎左冠狀動脈前降支復制MI模型,Sham組不進行結扎操作,其他手術程序和MI組保持一致。14 d實驗周期中,大鼠均腹腔注射給藥,具體給藥劑量為:SAB組:50 mg·kg-1·d-1,CID組:50 mg·kg-1·d-1SAB+40 μg·kg-1·d-1CID,Sham和MI組:等量生理鹽水。14 d后,處死大鼠,進行檢測分析。
2.2 HE染色將處死后大鼠的心臟剪取左心室,含室間隔部分,參照之前的方法[3],制成纖薄的組織切片,HE染色,厚度4 μm,置入普通顯微鏡的40×物鏡鏡頭下觀察。
2.3 Masson染色組織取材同HE染色。參照之前的方法[3],制成Masson染色切片。染色后,以膠原纖維為主的肉芽和瘢痕組織示藍綠色,心肌組織示紅色。膠原纖維區域大小用ZEN 2.3 lite軟件進行分析,其占左心室的比例用膠原容積分數(collagen volume fraction,CVF)值確定。
2.4 TUNEL染色組織取材同HE染色,按照試劑盒說明,石蠟切片,根據TUNEL染色操作步驟,并滴加DAPI復染,置入避光暗室中5 min,PBST清洗3次×1 min,封片。置入LSM 800激光共聚焦顯微鏡的40×物鏡鏡頭下觀察,計數染色陽性的細胞數/視野,取5個不重復視野/切片。
2.5 免疫組化(immunohistochemistry, IHC)組織取材同HE染色。參照之前的方法[3],將組織祛除干擾雜質后結合PKD1(1 ∶500稀釋)或HDAC7(1 ∶200)一抗,4 ℃冰箱中放置12 h,重復PBS沖洗3次×3 min,羊抗兔IgG二抗(1 ∶1 000)標記室溫15 min,DAB染色3 min。置入普通顯微鏡的40×物鏡鏡頭下,計數5個不重復視野染色陽性的細胞數的平均值。
2.6 免疫熒光(immunofluorescence, IF)組織取材同HE染色,切片脫蠟后微波處理15 min,PBS沖洗3次×3 min后兔血清封閉切片,加入一抗抗體PKD1(1 ∶50)或HDAC7(1 ∶50),4 ℃孵育過夜。d 2取出,PBST沖洗3次×3 min,和Cy3標記的熒光二抗共孵育1 h,滴加DAPI復染,封片。LSM 800激光共聚焦顯微鏡的40×物鏡鏡頭下觀察,ZEN 2.3 lite分析軟件計算紅藍熒光比值。
2.7 免疫印跡(Western blot, WB)用RIPA裂解緩沖液(添加蛋白酶抑制劑)浸泡冰箱中凍存的左心室心肌組織,制成組織勻漿后,離心分離提取總蛋白,BCA法確定總蛋白量。每份標本取40 μg蛋白行凝膠電泳,轉膜、封閉,孵育PKD1(1 ∶500)或HDAC7(1 ∶100)的一抗抗體,PBS沖洗3次×3 min后和羊抗兔二抗(1 ∶1 000)共孵育,持續2 h,Odyssey掃描儀掃描蛋白條帶。α-View SA軟件計算樣本蛋白/GAPDH參照蛋白的值。
3 結果
3.1 SAB對MI大鼠組織形態學的影響由Fig 1可見,相比Sham組大鼠完整的心肌細胞和組織結構,MI組大鼠心肌組織完整性受到嚴重破壞,以肉芽和瘢痕組織為主,間雜以殘存的肥大心肌細胞;SAB治療組大鼠完整的心肌和細胞較Sham組增多,少見肉芽和瘢痕組織;CID阻斷劑組大鼠心肌組織和細胞結構和MI組相似。這表明SAB可改善心梗后受損的心肌組織形態學。

Fig 1 Effect of PKD1 on histomorphology of MI rats (HE×400)
3.2 SAB對MI大鼠心肌纖維化的影響由Fig 2可見,Sham組大鼠心肌成分也有少量膠原纖維,呈網狀分布,以紅色心肌纖維為主; MI組大鼠藍色纖維,膠原占比較Sham組升高(P<0.01);SAB組大鼠心肌組織結構和Sham組接近,以紅色心肌組織為主,和MI組相比,膠原占比下降(P<0.01);CID組大鼠心肌組織結構和MI組接近,主要是肉芽和瘢痕組織,和SAB組相比,膠原占比升高(P<0.01)。這表明SAB可減少心梗后受損心肌組織的膠原占比。
3.3 SAB對MI大鼠心肌細胞凋亡的影響由Fig 3可見,Sham組大鼠心肌組織幾乎全部呈藍色熒光(示正常心肌細胞);MI組大鼠心肌組織中藍色和紅色熒光(示凋亡細胞)混合呈現,細胞凋亡計數較Sham組增加(P<0.01);SAB組大鼠心肌組織中主要呈現藍色熒光,間雜少量紅色熒光,細胞凋亡計數較MI組減少(P<0.01);CID組大鼠心肌組織中藍色和紅色熒光混合呈現,接近于MI組大鼠,細胞凋亡計數較SAB組增多(P<0.01)。這表明SAB可抑制因心梗后心肌組織受損導致的細胞凋亡。
3.4 SAB對MI大鼠心肌組織中PKD1和HDAC7蛋白表達的影響分別應用IHC染色(陽性表達蛋白示棕黃色)、IF染色(陽性表達蛋白示紅色熒光)和WB染色(陽性表達蛋白示灰黑色)檢測心肌組織中PKD1和HDAC7的蛋白表達。由Fig 4~7可見,三種檢測結果呈現一致的趨勢,均表明:Sham組大鼠的PKD1基礎表達量較高,HDAC7基礎表達量很低;與Sham組相比,MI組大鼠PKD1表達明顯降低(P<0.01),而HDAC7表達和Sham組基本一致;與MI組相比,應用了SAB后,PKD1和HDAC7的表達明顯升高(P<0.01),也高于Sham組(P<0.01);CID組和MI組基本一致,PKD1和HDAC7表達陽性的細胞數量均較SAB組明顯減少(P<0.01)。這表明SAB可上調心梗后心肌組織內PKD1和HDAC7的表達。

Fig 2 Effect of SAB on myocardial fibrosis in MI rats n=10)

Fig 3 Effect of SAB on cardiomyocyte apoptosis in MI rats n=10)

Fig 4 Effect of SAB on PKD1 and HDAC7 protein expression in myocardium of MI rats

Fig 5 Effect of SAB on PKD1 protein expression in myocardium of MI rats (IF)

Fig 6 Effect of SAB on HDAC7 protein expression in myocardium of MI rats (IF)

Fig 7 Effect of SAB on PKD1 and HDAC7 protein expression in myocardium of MI rats (WB) n=10)
4 討論
本研究大鼠組織形態學變化表明,MI組大鼠心肌梗死后缺血壞死的組織由肉芽組織或者瘢痕組織替代,后兩種組織的主要成分為膠原纖維,所以MI組大鼠CVF占比較高;并且心肌缺血受損后除了心肌細胞壞死外,還伴發著病理性凋亡,這也解釋了本研究MI組出現紅色熒光的細胞數明顯增多的原因。無論是壞死還是凋亡,心肌細胞的總量最終都會減少,又無法通過新的再生細胞的補充,剩余的功能細胞負荷加重,心肌細胞需要肥大來適應這一病理學進程,這是HE染色圖片中出現部分肥大細胞的原因,和文獻報道[7]的MI后殘存的心肌組織中有30%的心肌細胞出現肥大相一致。心肌細胞壞死、凋亡和病理性肥厚,如果不能采取有效的干預措施,將會出現心功能衰竭的嚴重后果[5, 8]。SAB給藥干預14 d后,大鼠心肌組織恢復較好,肉芽或者瘢痕組織明顯減少,CVF占比和凋亡細胞數量急劇下降,但這一藥理學作用可以被CID所阻斷。這表明,SAB可以保護MI后大鼠缺血損傷的心肌組織,而CID可以抑制SAB的保護作用。
PKD1和HDAC7的蛋白表達分別應用IHC染色、IF染色和WB染色三種不同方法進行檢測分析,檢測結果保持一致。正常的心肌組織中存在一定量的PKD1表達,但HDAC7的表達量很低。PKD1較高的基礎表達量可能與PKD1本身的生物學功能和生理學效應比較廣泛,表達位點較多相關[9],HDAC7的基礎表達量過低,可能是其正常情況下處于非活化狀態,需要PKD1激活并遷移至細胞核中,才能和其發生生物學偶合后磷酸化激活而發揮生物學效應[9]。MI后受損心肌組織中的PKD1較Sham組減少明顯,這也和心梗心肌的功能部分喪失密切相關;HDAC7的表達和Sham組接近,表明損傷并不誘發HDAC7的表達上調。
SAB用藥后,PKD1和HDAC7的表達均明顯上調,并且伴隨著缺血心肌組織受損的明顯減輕。之前的研究證實[5],PKD1用藥可以改善MI大鼠心肌組織的血流動力學參數,抑制膠原調控關聯蛋白,對抗心肌纖維化,減輕細胞凋亡,并能夠對抗缺血心肌組織中的產生的過度炎癥反應而保護心肌組織[8]。同時,PKD1是參與促血管新生的重要調控蛋白之一,可以直接上調VEGF的表達,促進毛細血管的新生,增加缺血心肌的微循環血供[6, 9-10]。VEGF反過來也可以通過VEGFR2-PKC信號通路激活PKD1,介導內皮細胞(endothelial cells,ECs)的增殖、遷移和管腔形成[9-10]。PKD1還具有介導內皮祖細胞(endothelial progenitor cells,EPCs)[4]和間充質干細胞(mesenchyma stem cell,MSCs)[11]增殖、遷移和管腔形成的作用,而EPCs和MSCs是誘導內源性血管生成的重要的細胞來源。HDAC7是PKD1的底物之一,可被VEGF介導的PKD1信號轉導刺激磷酸化激活后,由細胞核向細胞質遷移集聚,促進ECs的遷移、增殖和微血管的出芽新生[9, 12]。結合以上文獻報道和本研究結果表明,SAB可能通過調控PKD1/HDAC7軸在促缺血心肌組織血管新生,增加缺血心肌微循環血供,保護受損心肌中發揮著關鍵的作用。
本研究還發現,應用PKD1的特異性阻斷劑CID后,PKD1/HDAC7的表達明顯下調。這進一步表明,SAB很可能通過調控PKD1/HDAC7軸而保護缺血受損的心肌組織。
