基于CRISPR/Cpf1系統下的誘導型多基因激活體系的建立
2021-01-28 03:21:10黃秋月李中瀚殷旖珂
四川大學學報(自然科學版) 2021年1期
黃秋月, 李中瀚, 殷旖珂
(四川大學生命科學學院, 成都 610064)
1 引 言
細胞的譜系分化與命運決定一直是細胞生物學研究的重點方向.體細胞重編程與功能細胞間轉分化研究表明,哺乳動物細胞可通過特異性地表達少數幾個特征性的轉錄因子而實現細胞命運的相互轉化,例如Oct4、Sox2、Nanog和Lin28可將成纖維細胞重編程為誘導性多能干細胞(iPSC,induced pluripotent stem cells)[1];Ascl1、Brn2、Myt1l的表達可實現成纖維細胞向神經元轉換[2];而 Hnf4α、Foxa1/2以及Gata4、Mef2c、Tbx5組合則可以分別將成纖維細胞轉分化為肝實質樣細胞和心肌細胞[3-4].這些研究表明,通過激活特征轉錄因子的表達,可實現細胞命運與功能的人工改造與轉變.
傳統的基因過表達策略通常依賴質粒、慢病毒、人工mRNA等形式將含有強啟動子的轉基因引入靶標細胞中,但是此類方法的缺陷在于當需要引入多個基因時,投遞效率較低、載體能承受的基因大小受限以及外源基因整合可能破壞細胞內源基因及其調控元件等,因此,開發高效率的內源基因激活技術逐漸成為當前重編程與轉分化研究中的重點方向之一.隨著基因編輯技術的發展,尤其是CRISPR/Cas9系統的廣泛應用,利用核酸酶活性突變體dCas9及轉錄激活結構域構建靶向內源基因的基因激活技術已見報道,并被成功用于重編程與轉分化研究中,例如,可將MEF細胞重編程為iPSC[5]、成纖維細胞轉分化為骨骼肌細胞[6]、將iPSC定向分化為神經元等[7].然而,dCas9介導的多基因激活須將多個sgRNA轉錄元件串聯,且每個元件皆須具備U6啟動子、終止子等序列,造成質粒構建繁瑣且載體能承載的sgRNA元件數量有限.而近年來興起的CRISPR/Cpf1多基因編輯因其具有RNA酶活性,能夠實現pre-crRNA中多個crRNA的剪切和釋放[8-9],成為更具潛力和普適性的多基因激活工具.
Cpf1蛋白晶體采用識別葉 (Recognition lobe,REC) 和核酸酶葉 (Nuclease lobe,NUC)的二裂片結構,呈蟹鉗狀[10].根據Cpf1識別功能域和內切酶功能域分開的特點,將NUC進行定點突變使其失去核酸內切酶活力,并與轉錄激活域融合構建出基因激活工具.為了更能夠了解生命活動的情況,研究各生命機體生化反應下的多基因調控機制,本論文擬構建以CRISPR/Cpf1系統為基礎的誘導性基因激活系統.
基于此,本論文采用了在C端融合三元轉錄激活域VPR(VP64-P65-Rta)的dAsCpf1、dFnCpf1、dLbCpf1,篩選獲得了最強激活效果的dAsCpf1-VPR系統.此外,在該系統中還引入了不穩定結構域(DD-domain)和四環素控制的操縱子,從蛋白水平和轉錄水平分別控制融合蛋白表達,從而實現了特定時間、定向激活基因轉錄.最后,本論文實現了MYOD1、Oct4兩個基因同時激活,證明了TRE3G-DD-dAsCpf1-VPR系統具有誘導性多基因激活的能力.
綜上所述,在本論文中,我們成功建立了基于CRISPR/Cpf1系統的可誘導性的多基因激活體系,能夠快捷、高效的通過靶向DNA進行誘導內源性激活,在轉錄水平進行有效調控,從而具備定向調控細胞基因表達的效果,為實現細胞命運與功能的人工改造與轉變奠定技術基礎.
2 材料和方法
2.1 材料和試劑
人類胚胎腎細胞系HEK 293FT,小鼠胚胎成纖維細胞系NIH3T3,DMEM basic培養基(Gibco,C11995500BT),胎牛血清(Natocor,NTC-HK009),抗生素-抗真菌劑(Gibco,15240-062),轉染試劑Doge Fect (內蒙古尼迪生物,23427-01),SuperScriptTMⅡReverse Transcriptase試劑盒(Invitrogen,638315),細胞裂解液M-PER(Thermo scientific,QJ222436),GAPDH抗體(華安,R1208-3),HA抗體(Sigma,F1804-50 μg),Anti-rabbit二抗(Invitrogen,SA5-10044),Anti-mouse二抗(Invitrogen,SA5-10172),polybrene(Millipore,638313).
2.2 方 法
2.2.1 瞬時轉染 6孔板中每孔鋪7.5×105個細胞,24 h后,在顯微鏡下檢查細胞覆蓋率,當細胞覆蓋率達到60%~70%時進行轉染.轉染時,取6 μL Dogo加入94 μL DMEM中渦旋混勻后,瞬離并靜置5 min;同時1 μg目的質粒用DMEM補齊體積至100 μL;瞬離后加入靜止5 min后的Dogo混合液渦旋混勻,室溫靜置30 min;最后將脂質體與質粒混合液緩慢滴入細胞孔板中,輕搖混勻后放入37 ℃、5% CO2培養箱中培養.
2.2.2 免疫印跡 去除培養基后,加入預冷的PBS,用細胞刮刀收集細胞置于1.5 mL的EP管中,3000 r/min離心5 min,去上清,加入適量細胞裂解液M-PER (Thermo scientific),用移液器吹打均勻(冰上操作).每10 min震蕩一次,重復3次,隨后于4 ℃,14 000 g離心20 min,收集上清,加入4×蛋白上樣液,95 ℃孵育5 min使蛋白變性.SDS-PAGE電泳分離蛋白,23 V恒壓半干轉轉膜30 min.用溶于TBST的5%脫脂奶粉封閉1 h,一抗(Anti-GAPDH 1∶2 000,Anti-HA 1∶1 000)于4 ℃冰箱中低溫孵育過夜,二抗(Anti-rabbit 1∶10 000,Anti-mouse 1∶10 000)常溫孵育1 h,顯影.
2.2.3 熒光定量 去除培養基后,加入1×PBS清洗2~3次,Trizol法提取目的細胞的總RNA,使用Invitrogen SuperScriptTMⅡ Reverse Transcriptase試劑盒,將RNA反轉錄為cDNA.使用BIO-RAD SYBR Green Supermix試劑盒進行熒光定量分析,其中Gapdh為內參.
qPCR引物:
mus-Gapdh-F: GCACAGTCAAGGCCGAGAAT
mus-Gapdh-R:GCCTTCTCCATGGTGGTGAA
mus-Myod1-F:AGCGACACAGAACAGGGAAC
mus-Myod1-R:TCGAAAGGACAGTTGGGAAG
mus-Oct4-F:GCCCTCCCTACAGCAGATCACTCACATCG
mus-Oct4-R:AAGGTGTCCCTGTAGCCTCATACTCTTCTCGT
2.2.4 質粒構建
a) dAs/Fn/LbCpf1-VPR的構建
使用KOD-Plus Mutagenesis Kit(公司)點突變試劑盒分別將pcDNA3.1-As/Fn/LbCpf1-3×HA的908位、832位、917位的天冬氨酸突變為丙氨酸;在青蘭基因合成VPR序列,通過infusion的方法將VPR連接在dCpf1的C端,構建成pcDNA3.1-dAs/Fn/LbCpf1-3×HA-VPR ;
b) TRE3G-DD-dAsCpf1-VPR的構建
將pLVX-TRE3G空載酶切,將DD Domain片段PCR并純化,并將dAsCpf1-3×HA-VPR片段PCR并純化后,用infusion的方法將DD Domian連接于dAsCpf1-3×HA-VPR的N端,構建成pLVX-TRE3G-DD-dAsCpf1-3×HA-VPR;
3 結果分析
3.1 構建CRISPR/Cpf1-VPR基因激活系統
將來源于Acidaminococcussp.的AsCpf1的第908位,Francisellanovicida的FnCpf1的第917位和Lachnospiraceaebacterium的LbCpf1的第832位的天冬氨酸突變為丙氨酸,并且在dAs/Fn/LbCpf1的C端連接了由VP64,P65和Rta(VPR)蛋白組成的三元復合轉錄激活因子的激活效應區域(TAD),形成具有激活作用的融合蛋白(圖1a).將需激活的靶標基因的sgRNA設計在該基因轉錄起始位點(Transcription Start Site,TSS)之前,dCpf1-VPR在sgRNA的介導下能夠特異性識別并且結合在該基因啟動子或者增強子區域,VPR通過招募細胞因子或其他共轉錄激活因子,并結合形成多元復合物,進而定向激活靶標基因(圖1b).
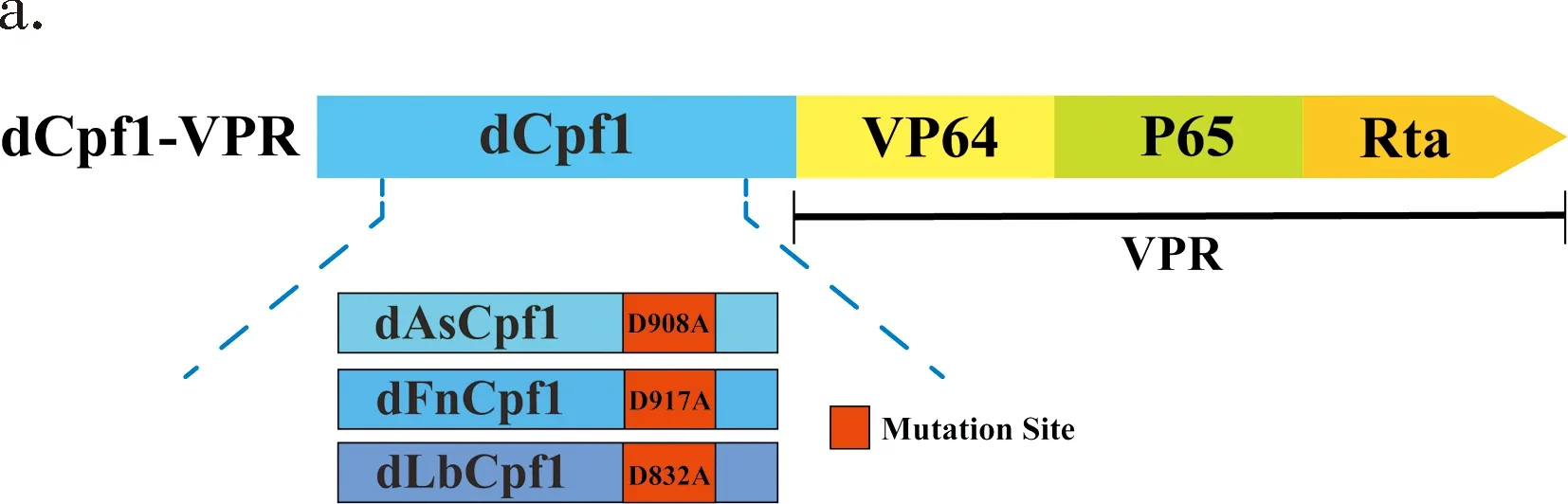
圖1 dCpf1-VPR激活系統的構建策略
3.2 dCpf1-VPR激活系統能夠定向上調基因的轉錄
圖2為NIH3T3細胞中Oct4基因的轉錄水平.結果顯示,dAs/Fn/LbCpf1-VPR三種激活系統都有不同程度的激活作用,其中在dAsCpf1-VPR系統下的Oct4基因mRNA水平與對照組相比可提升到104倍,證明dAsCpf1-VPR系統激活效果最強.因此我們將選用dAsCpf1-VPR激活系統進行后續的實驗. 同時測試由單個sgRNA介導下的激活效果,sgRNA-2和5介導的激活效果與對照組相比提升到102倍,并且證明了由sgRNA-2和5介導的激活效果與5個sgRNA介導的的強度相同,與對照組相比可提升到104倍.
3.3 構建誘導性激活系統
為了構建誘導性基因激活系統,將TET-ON系統和不穩定配體(DD Domain)與dAsCpf1-VPR相結合,形成可由TMP和Dox小分子藥物誘導的基因激活系統(圖3).DD Domain是來源于大腸桿菌二氫葉酸還原酶(DHFR)突變體的不穩定結構域,能夠在小分子化合物TMP的存在下保持結構穩定,促使其融合蛋白DD-dAsCpf1-VPR穩定表達.TET-ON系統中蛋白的表達受到TRE3G啟動子的控制.只有在Dox存在的情況下,rtTA才能促使TRE3G啟動子發揮作用并致使下游基因的轉錄.
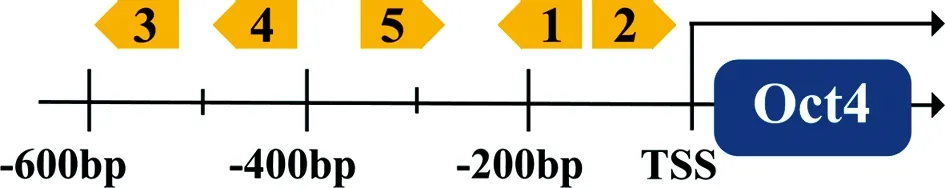
圖2 dCpf1-VPR系統定向激活Oct4基因轉錄情況

圖3 構建誘導性激活系統
3.4 TRE3G-DD-dAsCpf1-VPR系統實現蛋白穩定表達和誘導性基因激活
為了驗證TRE3G-DD-dAsCpf1-VPR系統的誘導控制效果,分別從蛋白水平和轉錄水平進行驗證.圖4a結果顯示,在Dox和TMP雙藥誘導下DD-dAsCpf1-VPR融合蛋白均得到高表達,且誘導系統的本底水平表達量很低.同時圖4b結果顯示,加雙藥誘導組與對照組相比,Oct4基因的mRNA水平提高了250倍,能顯著提高Oct4基因的轉錄水平.以上結果說明,TRE3G-DD-dAsCpf1-VPR系統可以在Dox和TMP雙藥的誘導下,實現對目標基因的誘導性激活.
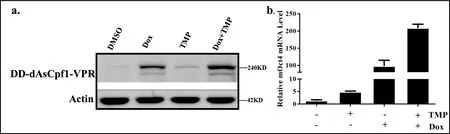
圖4 Dox和TMP雙藥誘導蛋白表達和基因激活情況
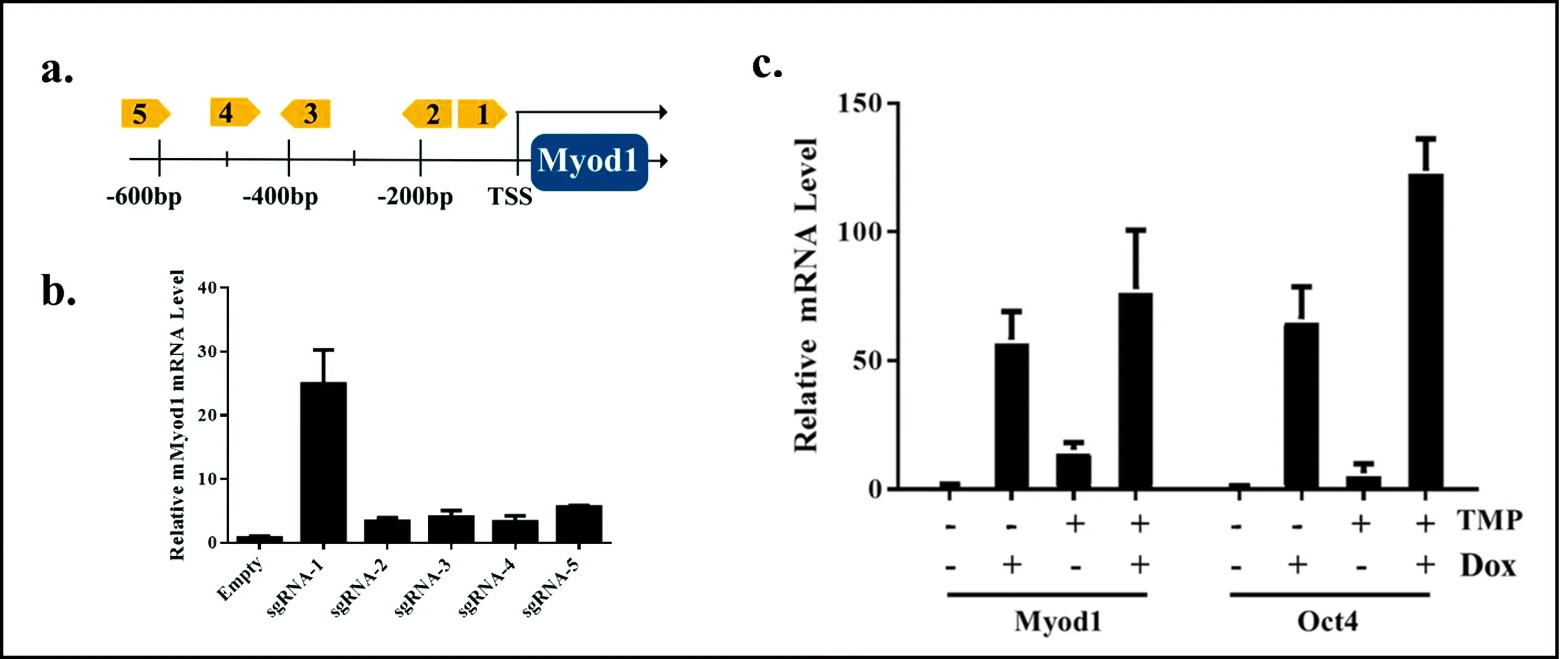
圖5 單個sgRNA介導Myod1基因的激活情況和Dox、TMP雙藥誘導下多基因激活的情況
3.5 TRE3G-DD-dAsCpf1-VPR系統能夠介導誘導性多基因激活
圖5b為Myod1基因由單個sgRNA介導的激活效果,篩選出了激活效果較強的sgRNA-1和5.然后同時對Myod1、Oct4基因進行激活,根據圖5c的結果顯示,Myod1、Oct4基因的轉錄水平分別在
Dox和TMP單藥的控制下,能夠實現不同程度的轉錄水平的提升.在Dox誘導下,Myod1和Oc4基因的轉錄水平分別提升了大約50倍和70倍,在同時加雙藥的條件下可分別提高80、120多倍,表明TRE-3G-DD-dAsCpf1-VPR系統可以高效介導誘導性多基因激活.
4 討 論
隨著基因編輯技術的發展,尤其是CRISPR/Cas9系統的廣泛應用,利用核酸酶活性突變體dCas9及轉錄激活結構域構建靶向內源基因的基因激活技術已見報道,并被成功用于重編程與轉分化研究中.然而,dCas9介導的多基因激活須將多個sgRNA轉錄元件串聯,且每個元件皆須具備U6啟動子、終止子等序列,造成質粒構建繁瑣且載體能承載的sgRNA元件數量有限.CRISPR/Cpf1系統是近年來興起的多基因編輯系統,其突出特點是,Cpf1具有RNA酶活性,能夠促使pre-crRNA中的多個crRNA的剪切和釋放,因此更具備作為多基因激活工具的潛力和普適性.
為了能夠更好地實現細胞命運與功能的人工改造與轉變,本文構建了基于CRISPR/Cpf1系統下能夠定時定向激活多基因的工具.為了篩選出最高效激活能力的Cpf1,對比了dAs/Fn/LbCpf1-VPR的激活效果.基于此,為了構建可誘導的Cpf1基因激活技術方法,在dAsCpf1-VPR的N端引入了不穩定結構域(DD-domain)和四環素控制的操縱子,通過在轉錄和蛋白穩定性兩個層面添加誘導性調控元件,證明能夠在Dox和TMP雙藥調控下蛋白水平和轉錄水平的高效誘導表達和激活.最后,對MYOD1、Oct4兩個基因同時激活,證明TRE3G-DD-dAsCpf1-VPR系統具有誘導性多基因激活的能力.綜上所述,成功將CRISPR/Cpf1系統與TET-ON系統融合,建立了誘導性多基因激活體系,能夠快捷、高效的通過靶向DNA進行誘導內源性激活,在DNA水平上對細胞轉錄水平進行有效調控,具有定向調控細胞基因表達,能夠實現細胞命運與功能的人工改造與轉變的潛力.
猜你喜歡
工業設計(2022年8期)2022-09-09 07:43:20
美與時代·美術學刊(2022年3期)2022-04-27 01:18:15
軍民兩用技術與產品(2021年10期)2021-03-16 06:05:30
北京測繪(2020年12期)2020-12-29 01:33:58
火花(2019年12期)2019-12-26 01:00:28
裝備制造技術(2019年12期)2019-12-25 03:06:46
人大建設(2019年12期)2019-05-21 02:55:32
中國洗滌用品工業(2019年4期)2019-05-11 09:27:34
家庭影院技術(2017年9期)2017-09-26 03:41:45
學苑創造·A版(2015年11期)2016-01-14 09:03:27
