伴有t(6;9)(p23;q34)/DEK-NUP214融合基因陽性的急性髓系白血病患者的特征分析*
2021-02-23 08:47:34李麗寧郭華燕張艷敏舒汨汨
檢驗醫學與臨床 2021年4期
董 瑩,李麗寧△,郭華燕,張艷敏,張 琴,雷 鑫,閆 芳,徐 莉,舒汨汨
1.西北大學附屬醫院/西安市第三醫院血液病與腫瘤中心,陜西西安 710018;2.西北大學附屬醫院/西安市第三醫院輸血科,陜西西安 710018;3.中國人民解放軍空軍軍醫大學第一附屬醫院/西京醫院血液內科,陜西西安 710032
急性髓系白血病(AML)是一種造血干細胞惡性增殖克隆性疾病,目前發現其發病與染色體易位形成新的融合基因有關,其中6號染色體6p23的DEK基因與9號染色體9q34的NUP214基因重排,形成DEK-NUP214融合基因,產生DEK-NUP214融合蛋白,又名DEK-CAN蛋白。t(6;9)(p23;q34)是一種造血系統罕見的染色體異常,該染色體易位發生率很低,在成人AML中的發病率為0.7%~1.8%[1-3]。2008年WHO已將伴有t(6;9)(p23;q34)的AML添加到造血與淋巴組織腫瘤分類系統中,作為一種獨立的AML臨床亞型[4],此類疾病預后極差,復發風險高,應該在早期緩解后盡快行骨髓移植。由于這種染色體易位極為罕見,目前對它的認識僅僅來源于一小部分患者,對它的認知仍十分有限,其致病機制也不清楚,還有待進一步研究。本研究對2013年1月至2018年12月在中國人民解放軍空軍軍醫大學第一附屬醫院/西京醫院血液內科確診的8例伴有t(6;9)(p23;q34)/DEK-NUP214融合基因陽性的AML患者進行回顧性分析,旨在進一步認識此類疾病的臨床特征,探討其治療策略,加強對此類疾病的規范化管理。
1 資料與方法
1.1一般資料 8例AML患者均為2013年1月至2018年12月在中國人民解放軍空軍軍醫大學第一附屬醫院/西京醫院血液內科收治的初發病例,其中男6例,女2例;年齡15~66歲,中位年齡48歲。所有病例均按照法、美、英協作組(FAB)標準和WHO血液系統腫瘤標準,經骨髓形態學、白血病免疫分型、細胞遺傳學及分子生物學檢測確診。
1.2方法
1.2.1免疫分型分析 取肝素抗凝骨髓4 mL,采用常規四色免疫熒光法對8例患者進行免疫表型分析。所用單克隆抗體包括CD2、CD7、CD20、CD10、CD34、HLA-DR、CD117、CD11b、CD14、CD13、CD19、CD33、CD56、CD64、CD61、CD41、cCD79、cMPO、cCD3、CD235a、CD71、CD138、CD15,用流式細胞儀(COULTER EPICS XL,美國貝克曼-庫爾特公司)和CXP2.0軟件獲取并分析10 000個細胞,通過CD45/側向角散射(SSC)設門法識別白血病細胞群,再分析計算該細胞群各相關抗原的表達,分析時以原始幼稚細胞群中抗原表達>20%作為陽性。
1.2.2染色體制備和核型分析 抽取患者初次就診時的骨髓細胞,24 h短期培養后收獲細胞,并加入秋水仙酰胺使分裂的細胞停止在分裂中期,按要求制備染色體標本,應用G顯帶技術進行染色體核型分析。核型異常命名根據《國際人類細胞遺傳學命名法(ISCN)2005》進行。
1.2.3實時熒光定量PCR 取乙二胺四乙酸二鈉(EDTA-Na2)抗凝的患者骨髓標本2 mL,用紅細胞裂解液裂解紅細胞,1 500 r/min離心10 min,離心得到白細胞沉淀,Trizol經典法提取骨髓細胞總RNA。RNA反轉錄合成cDNA,以cDNA為模板,用急性白血病43種融合基因檢測試劑盒(上海源奇生物醫藥科技有限公司)檢測患者是否含有特異性融合基因,檢測的基因包括BCR-ABL1、RUNX1-RUNX1T1、CBF-MYH1、PML-RARa、DEK/NUP214、SIL/TAL1、E2A/HLF、TEL/AML1、E2A/PBX1、NMP/MLF1、TEL/PDGFRB、TLS-ERG、SET/CAN、ETV6/PDGFRA、FIP1L1/PDGFRA、NPM/ALK、RARa相關基因、TEL相關基因、MLL相關基因、NUP98相關基因、AML1相關基因等。
1.2.4骨髓細胞DNA提取及AML相關基因突變檢測 取EDTA-Na2抗凝的患者骨髓標本2 mL,用血液基因組DNA提取試劑盒(北京天根生物技術有限公司)提取總DNA。用一代測序儀(3500Dx,ABI公司)檢測FLT3-TKD、FLT3-ITD、NPM1、CEBPA、C-Kit 8、C-Kit 17基因突變。
2 結 果
2.1血常規和骨髓細胞形態學特點 8例患者均伴有外周血白細胞計數升高,白細胞計數為(12.35~114.61)×109/L,血紅蛋白為46~99 g/L,血小板計數為(18~160)×1012/L。根據患者的臨床表現、血常規、骨髓形態學、骨髓病理學和細胞化學染色結果,并參照免疫分型結果,按照FAB標準分型,臨床診斷如下:AML未分化型M1 1例,AML部分分化型M2 5例,急性粒-單核細胞白血病M4 2例。
2.2免疫分型結果 根據CD45熒光強度與SSC把病例中的骨髓有核細胞區分成3群細胞,包括異常細胞群、正常粒細胞群、正常淋巴細胞群。8例患者中,主要表達HLA-DR(7/8)、cMPO(7/8)、CD33(8/8)、CD13(8/8)、CD34(6/8)、CD11b(4/8)、CD117(7/8),未發現染色體核型,基因與免疫表型之間有明顯關聯。
2.3染色體核型檢測結果 對8例AML患者進行染色體核型分析,觀察分析250個分裂中期細胞,其中2例沒有分裂相,剩余6例中20個中期分裂相的所有細胞均顯示t(6;9)(p23;q34)染色體。
2.4基因檢測結果 8例AML患者進行43種急性白血病融合基因檢測,結果均檢出DEK-NUP214融合基因陽性(附其中1例結果見圖1)。其中4例進行基因突變(FLT3-TKD、FLT3-ITD、NPM1、CEBPA、C-Kit 8、C-Kit 17)檢測,2例檢出FLT3-ITD突變陽性。
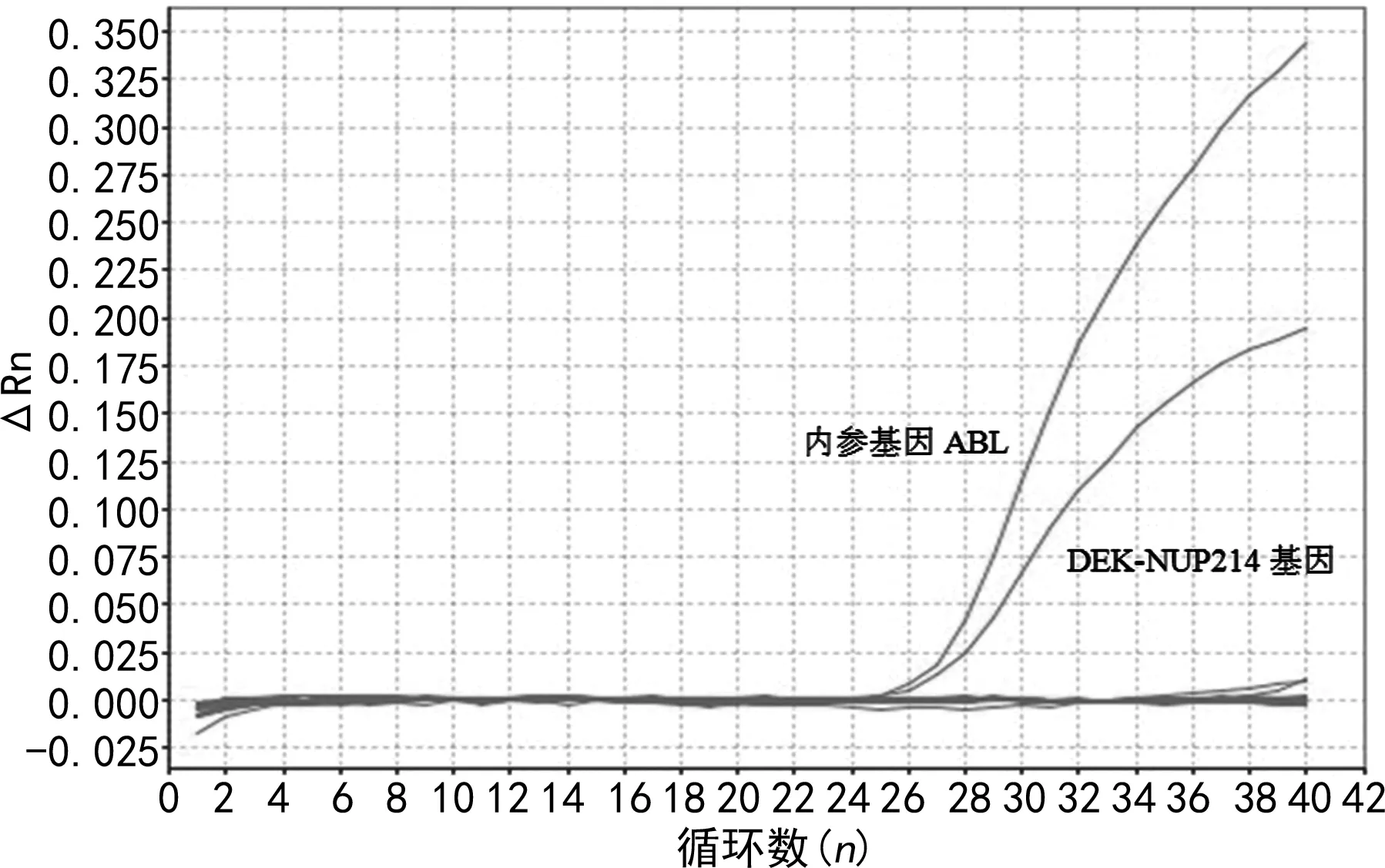
圖1 實時熒光定量PCR檢測AML患者DEK-NUP214融合基因陽性的擴增曲線
2.5臨床治療及預后 對8例AML患者治療情況進行分析,其中3例未在中國人民解放軍空軍軍醫大學第一附屬醫院/西京醫院治療;1例入院后未行化療即死亡;剩余4例患者,2例先用小劑量EA預處理,再行標準劑量IDA方案化療,1例患者化療2個療程未緩解死亡,1例患者化療3個療程后死于感染,剩余2例行地西他濱聯合CAG方案化療,鞏固4個療程后,1例緩解后轉上級醫院行異基因造血干細胞移植術,術后1.5年因肺部感染死亡,1例接近緩解(原始粒細胞接近5%),但是很快復發,再更換化療方案治療,治療無效死亡。
3 討 論
血液系統疾病在病程發生過程中,會出現某些非隨機性染色體異常,這些染色體的相互易位形成特定的融合基因,最終表達生物學特性異常的融合蛋白,這些蛋白與血液病的發生關系密切[2,5-6]。伴t(6;9)(p23;q34)的AML是一類性質獨特、預后極差的疾病,此類疾病以年輕患者居多,發病率低,化療效果差,緩解率低,復發風險高,生存期短[2]。本研究報道的8例患者中免疫分型都表達CD13、CD33,與文獻報道類似[2,7-8]。盡管有報道稱,伴t(6;9)異常的AML患者骨髓形態學檢查易出現骨髓病態造血或者嗜堿性粒細胞增多[3,9],但是中國人民解放軍空軍軍醫大學第一附屬醫院/西京醫院確診的8例患者,其中2例患者出現病態造血,8例患者骨髓形態學檢查均未觀察到嗜堿性粒細胞增多,分析可能與收集的病例數量有限,或者疾病的異質性有關。YOSHIMOTO等[10]發現,在青年AML患者中,FLT3-ITD基因的突變率為20%~30%,FLT3-TKD基因的突變率為7%,STONE等[11]、ALSABEH等[8]和OYARZO等[7]總結出這些基因在AML伴t(6;9)患者中的突變率為71%~88%,且基因突變與患者預后不良有關。本研究中有4例患者進行預后相關基因突變檢測,FLT3-ITD的檢出率為50%(1例為未化療即死亡,1例為化療2個療程未緩解死亡),因為收集到的病例有限,無法進行統計分析。由于伴有t(6;9)(p23;q34)的AML患者預后差,且患者緩解期短暫,因此,獲得完全緩解后的患者宜盡早進行異基因造血干細胞移植,以期獲得長期生存的機會。
伴t(6;9)(p23;q34)的AML代表一類獨特的AML亞型,盡管其分子生物學研究取得了一些進展,但目前,對DEK-NUP214融合基因和其產生的蛋白在腫瘤發生中的作用及臨床意義了解得很少,針對此類患者的治療方案仍處于研究階段,有研究指出尋找到針對DEK-NUP214融合基因的靶向治療藥物可能是此類疾病治療的突破口和轉折點[12-13]。
猜你喜歡
中學生數理化·中考版(2022年8期)2022-06-14 06:55:24
今日農業(2021年19期)2022-01-12 06:16:36
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中老年保健(2021年11期)2021-08-22 03:15:44
中學生數理化(高中版.高考數學)(2021年1期)2021-03-19 08:28:38
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
無線電工程(2020年11期)2020-10-29 01:25:46
