1例新生兒常染色體顯性智力障礙21型病例報告
2021-03-18 14:00:42陳蘭赫紋劉玲
中國當代兒科雜志 2021年3期
關鍵詞:新生兒
陳蘭 赫紋 劉玲
(貴陽市婦幼保健院新生兒科,貴州貴陽 550003)
患兒男,1日齡,因哭聲弱1 d,反復唇周青紫2 h入院。患兒系第2胎第2產,39+4周順產出生,1 min、5 min Apgar評分均為10分,出生體重3 100 g,出生史無特殊。母孕期定期產檢,孕23周時外院胎兒B超示后顱窩池略寬,約9 mm。后于我院行MRI檢查示胎兒后顱窩池略寬,前后徑最寬處約10.1 mm,行無創DNA檢查無異常。否認家族遺傳病史。其兄1歲10個月,體健。
入院體格檢查:T 36.4℃,P 145次/min,R 42次/min,體重 2.83 kg,血壓 66/40 mm Hg,身長49 cm,頭圍33 cm,胸圍32 cm;神志清楚,反應正常,營養中等,膚色紅潤,哭聲弱,哭時伴口唇發紺,呼氣相口唇呈“噗噗”吹氣樣表現。枕部突出,眼距寬,眼裂小,雙眼向外下斜,高腭弓,雙手通貫掌,右足扁平、外翻。余無特殊。入院胸片示新生兒肺炎、心臟增大。血常規檢查示WBC 15.76×109/L(參考值:5.0~21.0×109/L),血紅蛋白172 g/L(參考值:130~200 g/L),血小板 198×109/L(參考值:125~350×109/L),L 11.4%(參考值:20.0%~40.0%),N 81.3%(參考值:50.0%~70.0%);C反應蛋白(CRP)38.2 mg/L(參考值:0~5.0 mg/L)。肝腎功能、電解質正常。心臟超聲示先天性心臟病:動脈導管未閉(2.3 mm);房間隔缺損Ⅱ孔型(2.5 mm)。肝膽胰脾、雙腎B超無異常。頭顱MRI檢查示胼胝體壓部略顯短;大枕大池;少許蛛網膜下腔出血。染色體核型:46 XY。
入院后予低流量鼻導管吸氧,以及頭孢噻肟鈉抗感染治療。7 d后,復查感染指標均正常。入院后予泵奶喂養逐漸過渡至人工喂養。患兒吸吮力弱,喂養時伴有口唇發紺,予吸氧可緩解。入院7 d后患兒家長自行要求出院。
患兒出生有特殊面容,喂養困難,其臨床表現不能用感染、缺氧等原因進行解釋,為了盡快明確診斷,在征得患兒家長同意后,于出院前采集患兒及父母外周血各2 mL,送至第三方醫學檢驗所檢測(北京康旭醫學檢驗所有限公司),進行染色體拷貝數變異與全外顯子測序分析。
提取患兒及家系外周血中的基因組,通過低深度高通量測序對患兒進行染色體拷貝數分析,未見異常。使用覆蓋39 Mb人類基因組的IDT探針(IDT xGen Exome Research Panel包含約430 000個單獨合成的探針)對患兒外周血的基因組進行靶向區域捕獲測序。通過數據處理,獲得靶向區域突變位點信息,使用PolyPhen、SIFT、Mutation Taster軟件對原始數據進行蛋白質功能損傷分析。根據患兒臨床癥狀、基因遺傳方式、變異頻率、測序深度等信息最終選出需要進一步驗證的突變位點。對突變位點的致病性按照美國醫學遺傳學與基因組學學會(ACMG)遺傳變異分類標準與指南進行分析和注釋。對于疑似突變位點,對患兒及家系采用Sanger測序進行驗證。
高通量全外顯子組測序分析顯示患兒CTCF基因雜合缺失4 bp,即c.778_781delAAAG。對該位點使用SIFT Indel算法,缺失的效果是“有害的”,因為它會導致第260位后氨基酸發生移碼,同時在移碼后的第2位引入終止密碼,使編碼的CTCF蛋白為截斷性蛋白(p.Lys260ValfsTer2),這意味著生成的CTCF只有它的N-末端區域,而缺少其11個鋅指結構域和C末端區域。對父母進行CTCF測序未發現攜帶這種突變。見圖1。
經 Sanger 測序驗證,患兒父母均未攜帶該變異,該變異為新發突變。結合患兒臨床表現及遺傳學分析結果,患兒被診斷為常染色體顯性智力障礙21型。患兒生后6月齡因喂養困難及反復感染夭折。
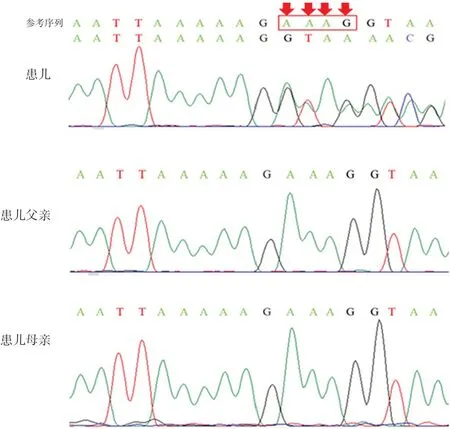
圖1 患兒及患兒父母CTCF Sanger測序峰圖 箭頭所示為CTCF c.778_781位點AAAG雜合缺失位置。
討論:Gregor等[1]2013年首次報道了CTCF(CCCTC結合因子;MIM 604167)突變與患者不同嚴重程度的智力殘疾、小頭畸形和生長發育遲緩有關。以“CTCFgene mutation”和“CTCF基因”為關鍵詞,從PubMed、中國知網、萬方等數據庫進行檢索,檢索時間自2013年1月至2020年4月,發現由CTCF基因突變致病或可能致病的患者的臨床詳細信息涉及49例(包含染色體拷貝數變異),其中39例為新生突變。49例患兒中,3例為中國患兒,分別為1例1歲7個月女童攜帶c.615_618delGAAA(p.Lys206Profs*15),1例7歲11個月男童攜帶c.329dupT(p.Gly111fs*29),1例8歲7個月女童攜帶c.1699C>T(p.Arg567Trp)[1-5]。少數病例未被檢索到詳細的臨床特征,不包括在這49例患兒中。如Meng等[6]應用全外顯子組測序對出生100 d左右的危重癥新生兒進行輔助診斷,其中1例29 d男性患兒為CTCF基因c.1103G>T(p.R368L)雜合新生突變,其父母未見變異;Willsey等[7]對不同地區511個復雜神經發育障礙的抽動穢語綜合征患兒進行全外顯子組測序數據分析,發現1例男性患兒為CTCF基因c.1244G>A(p.R415Q)突變,其父母未見變異;Retterer等[8]分析了3 040例包含各系統發育異常患兒的全外顯子組測序數據,發現2個神經系統異常的患兒分別存在CTCF基因c.1288_1298del11(p.H430Cfs*10)和c.1028A>G(p.Y343C)的變異位點,未明確是否為新發突變。本例患兒存在CTCF基因c.778_781delAAAG(p.Lys260ValfsTer2)雜合缺失,而未發現其父母攜帶這種突變,為新發突變。
在上述具有臨床詳細信息的49例患兒中,CTCF基因突變數據共計33個,錯義突變20個,無義突變2個,插入或缺失9個,剪切突變2個,還有5個在16q21處包含CTCF基因缺失的不同染色體變異,如1.5 Mb deletion、0.58 Mb deletion、1.1 Mb deletion、1.2 Mb deletion、0.28 Mb deletion。有約37%的患兒出現宮內發育遲緩,78%的患兒出現喂養困難,27%的患兒出現身材矮小,52%的患兒出現小頭畸形,79%的患兒出現智力障礙,77%的患兒出現行為異常,10%的患兒出現癲癇,26%的患兒出現聽力障礙,61%的患兒出現視力障礙,45%的患兒出現反復感染[1-5]。
CTCF基因位于16q22.1,約76.8 kbp,由13個外顯子組成,編碼727個氨基酸,是具有11個高度保守的鋅指結構域與約46個功能性的核酸結合位點的轉錄調控因子。近年來的研究發現,CTCF是脊椎動物體內的關鍵調控蛋白之一,參與細胞中的多種生物學過程,如基因轉錄的抑制與激活[9-11]。CTCF參與保護常染色質區,防止異染色質區域的擴展,進而阻斷轉錄增強子的調控起到絕緣子功能[12]。參與印記基因表達調控[13-14],也可影響DNA復制,如CTCF結合于母源等位基因ICR處,使其DNA復制時間較父源等位基因延后[15]。CTCF參與X染色體失活,可影響mRNA可變剪接[16-17]與染色質重塑[18-19]。
本例患兒與已報道CTCF基因突變患兒的部分臨床表型有不同程度的吻合[2-3]。如攜帶CTCF基 因c.688delG、c.773_776del、c.804_805delTG、c.782-2A>G、c.782-1G>T和c.329dupT的患兒均有輕度上瞼下垂、眼距寬、眼裂小、薄嘴唇、小下頜、高腭弓等面部畸形;攜帶CTCF基因c.329dupT和c.782-1G>T的患兒出生時有喂養困難;攜帶CTCF基因c.782-1G>T、c.329dupT和c.804_805delTG的患兒合并動脈導管未閉和/或房間隔缺損心臟畸形;攜帶CTCF基因c.688delG、c.773_776del、c.804_805delTG和c.782-2A>G的患兒合并有通貫掌或足部畸形;攜帶CTCF基因c.773_776del、c.782-2A>G、c.782-1G>T和c.329dupT的患兒有宮內發育遲緩。本例患兒雖未出現宮內發育遲緩,但出生后有喂養困難、面容異常、先天性心臟病、手足異常、胼胝體發育不良等臨床表型。因此,結合患兒臨床表現和基因檢測結果,患兒被確診為常染色體顯性智力障礙21型。
綜上所述,CTCF基因突變可導致認知和成長的可變損害,高通量測序技術有助于提高對新生兒罕見疾病的識別與診斷。本病例提示,對于臨床上有不明原因喂養困難,不能用感染、缺氧等原因進行解釋的新生兒,應盡早進行遺傳學分析,以幫助早期診斷和遺傳咨詢。
猜你喜歡
中國典型病例大全(2022年12期)2022-05-13 18:24:49
中華養生保健(2020年10期)2021-01-18 06:45:20
中華養生保健(2020年8期)2021-01-14 01:13:30
家庭醫學(下半月)(2019年9期)2019-10-12 08:04:06
家庭醫學(下半月)(2019年8期)2019-09-25 09:02:00
媽媽寶寶(2017年3期)2017-02-21 01:22:12
罕少疾病雜志(2016年4期)2016-03-11 16:34:39
中國衛生標準管理(2015年16期)2016-01-20 09:26:29
中國衛生標準管理(2015年6期)2016-01-14 05:17:08
中國衛生標準管理(2015年8期)2015-01-26 18:08:35
