先天性腎病綜合征1例報告及文獻復習
2021-03-25 02:24:10陳麗穎李志鑫劉振球
醫學理論與實踐 2021年6期
關鍵詞:基因突變
陳麗穎 李志鑫 饒 睿 劉振球
四川省樂山市人民醫院兒科 614000
先天性腎病綜合征(CNS)通常指出生3個月內發病,臨床表現符合腎病綜合征(NS,大量蛋白尿、低白蛋白血癥、嚴重水腫和高膽固醇血癥)。現將收治的1例CNS患兒的臨床表現、實驗室檢查、治療經過、基因分析等進行總結,并復習相關文獻,提高對該病的認識。
1 臨床資料
1.1 一般資料 患兒女,日齡13d。因“雙下肢浮腫、發現蛋白尿5+d”入院。患兒系第5胎,第2產,孕37+2周,單胎,因母親妊娠合并急性胰腺炎行剖宮產,Apgar評分不詳,產重3 320g,羊水Ⅲ度糞染,量30ml,臍帶、胎盤無異常,否認宮內窘迫,無胎膜早破,無窒息史。生后人工喂養,體重無明顯增長。家族史:患兒父親35歲,母親37歲,母親有胃炎、胰腺炎、高血脂病史,父親身體健康,非近親結婚,有一姐,8歲,腦癱。家人無類似病史,家族中無腎臟病患者。
1.2 檢查 入院查體:體溫36.5℃,脈搏132次/min,呼吸47次/min,血壓95/53mmHg(1mmHg=0.133kPa),體重3.38kg,身長52cm。外貌成熟兒,反應欠佳,面色、膚色蒼白,雙眼瞼輕度水腫,雙肺聽診呼吸音稍粗,未聞及干濕啰音。心率132次/min,心音欠有力,律齊,可聞及Ⅱ/Ⅵ級心臟雜音。腹膨隆,腹壁腫脹,無腹壁靜脈曲張,肝右肋下2cm,脾臟肋下未觸及,無移動性濁音,雙下肢腫脹壓之凹陷,會陰部腫脹。實驗室檢查:當地醫院實驗室檢查:(1)尿蛋白3+,潛血2+ ;(2)血生化:ALB 17.5g,腎功無明顯異常。(3)腹部彩超:肝膽胰脾雙腎及腹腔未見明顯異常。(4)胸片:右肺滲出性病變。入院后實驗室檢查:(1)尿液:黃色清亮,潛血1+,蛋白質3+,微量白蛋白定性(+)。(2)入院時查:總蛋白27g/L,白蛋白13.1g/L,血Ca2+1.62mmol/L 。膽固醇7.79mmol/L,甘油三酯4.05mmol/L。出院前查:總蛋白26.6g/L,白蛋白9.3g/L。(3)肌酐3.8μmol/L,尿微量蛋白濃度2.126g/L。(4)免疫Ⅰ號:免疫球蛋白G 0.884g/L,補體C3 0.48g/L,補體C4 0.11g/L。(5)HIV抗體、梅毒抗體、梅毒快速血漿反應素、結核抗體:陰性。(6)腹部彩超:①雙腎形態結構及血供未見明顯異常;②雙側輸尿管未見明顯擴張。(7)胸片:肺炎。(8)因患兒系新生兒,未行腎臟穿刺活檢。
1.3 治療及轉歸 患兒有大量蛋白尿,有嚴重低蛋白血癥,最低時白蛋白<10g/L,水腫,高膽固醇血癥。住院后反復給予人血白蛋白輸注,并給予補鈣、人免疫球蛋白等,經治療患兒白蛋白未提升,還有下降趨勢。入院治療17d后,家長放棄治療出院。
1.4 隨訪 出院時患兒仍存在水腫,出院前最近一次查白蛋白為9.3g/L,患兒于生后4個月左右死亡。
2 基因分析
患兒家系調查未發現家族中有類似疾病的成員。在知情同意情況下,采患兒、父、母的血完善先天性腎病相關基因檢測:發現患兒存在NPHS1p.R1109X(c.3325C>T),NPHS1p.D310N(c.928G>A)(剪切區)復合雜合突變。如圖1所示。

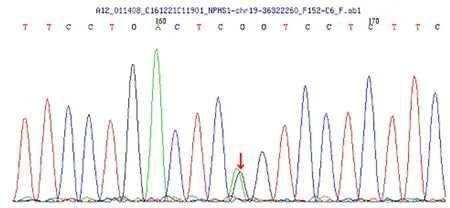
圖1 患兒基因檢測
發現患兒父親NPHS1基因存在雜合突變,c.3325C>T,(編碼區第3 325號核苷酸由胞嘧啶變異為胸腺嘧啶),導致氨基酸改變p.R1109X(無義突變),患兒母親無該位點變異。該變異不屬于多態性位點,在人群中發生頻率極低。見圖2。


圖2 患兒父親基因檢測
發現患兒母親NPHS1基因存在錯義突變,c.928G>A,(編碼區第928號核苷酸由鳥嘌呤變異為腺嘌呤),導致氨基酸改變p.D310N(第310號氨基酸由天冬氨酸變異為天冬酰胺)。患兒父親無該位點變異。該變異不屬于多態性位點,在人群中發生頻率極低。如圖3所示。


圖3 患兒母親基因檢測
3 討論
3.1 CNS的分型和基因突變 CNS根據病因可分為原發性(遺傳性)和繼發性(非遺傳性),繼發性由多種病原體宮內感染或母親疾病等導致,如梅毒、弓形體、巨細胞病毒、風疹病毒、肝炎病毒、人類免疫缺陷病毒以及瘧疾等,除感染外,母親SLE也可合并CNS[1]。原發性CNS由多個基因突變所致,目前已明確可以導致CNS的基因有NPHS1、NPHS2、WT1、PLCE1、LAMB2等[2],其中NPHS1、NPHS2編碼蛋白多為腎小球裂孔膈膜蛋白分子,WT1、LMX1B編碼蛋白為正常足細胞功能和發育所必需的轉錄因子或酶,LAMB2編碼蛋白為腎小球基底膜結構分子[3]。本例患兒基因檢測發現存在NPHS1基因雜合突變。本病發病機理為腎小球裂孔隔膜蛋白的編碼基因NPHS1突變導致,導致足細胞足突間的裂孔隔膜受損,從而出現大量蛋白尿。
依據有無其他系統受累,又可分為非綜合征型(non-syndromic)或單發型(isolated)和綜合征型(syndromic),前者包括:Nephrin基因突變(NPHS1、芬蘭型CNS)、Podocin基因突變(NPHS2)、WT1基因突變(單發型CNS)、PLCε1基因突變和LAMB2基因突變(單發型CNS);后者包括:WT1基因突變(Denys-Drash和Frasier綜合征)、LAMB2基因突變(Pierson綜合征)、LAMB3基因突變(赫茨交界性大皰性表皮松解癥)、LMX1B基因突變(甲—髕綜合征)、線粒體疾病(COQ2、COQ6)、CNS伴腦部畸形、Galloway Mowat綜合征(基因不明)、CNS伴其他畸形(沒有腦部畸形)和CNS伴心室擴大(基因不明)[4]。WT1基因突變為常染色體顯性遺傳,NPHS1、NPHS2、LAMB2和PLCε1等基因突變均為常染色體隱性遺傳[5]。
國外、國內報道CNS較常見的基因突變為編碼nephrin蛋白的NPHS1基因的突變,NPHS1基因突變導致的CNS綜合征,臨床上多指芬蘭型CNS綜合征,目前發現的突變位點數以百計,其中有2個典型突變:Fin-major(p.IJ41fsx91)和Finminor(p.Rll09x),94%的CNS病例有此兩型突變,前者占78%,后者為16%[6]。
3.2 CNS的治療 對于繼發性CNS,主要針對其原發病治療,如感染所致者,采用強有力的抗感染治療,病情常明顯好轉,不出現不可逆性腎臟損害[7]。對原發性CNS,無特殊有效的治療,類固醇激素或免疫抑制劑治療無效。主要采取對癥治療及營養支持治療。對于大多數原發性CNS患兒,腎移植是唯一有效的治療方式。預后差,病情進行性發展,多死于感染、腎衰竭、出血等。
猜你喜歡
英語世界(2023年6期)2023-06-30 06:29:10
中國醫學影像學雜志(2021年6期)2021-08-13 08:43:36
中國生殖健康(2020年2期)2021-01-18 02:51:26
小學生導刊(2018年13期)2018-06-29 03:49:00
中國生殖健康(2018年2期)2018-01-12 13:57:51
現代檢驗醫學雜志(2016年4期)2016-11-15 02:01:14
中國現代醫學雜志(2015年26期)2015-12-23 11:04:22
鄭州大學學報(醫學版)(2015年2期)2015-02-27 14:50:44
中華皮膚科雜志(2014年4期)2014-12-19 12:55:49
中國神經精神疾病雜志(2014年1期)2014-03-01 03:23:22
