固相萃取/液相色譜-質譜/質譜法測定山銀花中5種主要黃酮苷元的含量
2021-03-25 02:48:50史穎珠侯建波姚濱濱胡曉莉
分析測試學報 2021年3期
關鍵詞:黃酮
史穎珠,侯建波,謝 文,姚濱濱,盛 濤,胡曉莉,張 輝
(1.浙江大學 生物系統工程與食品科學學院,浙江 杭州 310058;2.杭州海關技術中心,浙江 杭州 310016;3.浙江省檢驗檢疫科學技術研究院,浙江 杭州 310016;4.浙江立德產品技術有限公司,浙江 杭州 310016)
山銀花為忍冬科植物灰氈毛忍冬(LoniceramacranthoidesHand.-Mazz.)、紅腺忍冬(LonicerahypoglaucaMiq,)、華南忍冬(LoniceraconfusaDC.)或黃褐毛忍冬(LonicerafulvotomentosaHsu et S.C.Cheng)的干燥花蕾或帶初開的花,并在夏初花開放前采收獲得。自《中國藥典》2005年版起,與金銀花進行區分。山銀花具有清熱解毒,疏散風熱的功效,可用于治療癰腫疔瘡、喉痹、丹毒、熱毒血痢、風熱感冒、溫病發熱[1]。研究發現其還具有抗菌、抗病毒、抗氧化和抗動脈粥樣硬化等作用[2-5],在我國主要分布在長江以南的湖南、四川、廣東、貴州、廣西、云南等省區和長江以北的河南、山東、安徽和湖北等省份[6]。
山銀花的有效成分除了綠原酸、灰氈毛忍冬皂苷乙和川續斷皂苷乙外,還有黃酮類、揮發油類、有機酸類和三萜類化合物[7-10]。有研究人員通過HPLC-MS/MS、光譜技術測定和鑒別山銀花黃酮類、有機酸類和環烯醚萜類化合物的含量[11-13],以及開展包含黃酮類化合物在內的指紋圖譜研究[14-16]。由于黃酮類化合物種類較多,在植物體內苷元可與糖類物質形成不同的苷而普遍存在[17-19],且標準品較難獲取,目前除液相色譜-高分辨質譜法[20-22],其他分析手段較難獲得更全面的信息。本文通過文獻調研篩選出山銀花中5種主要黃酮苷元及其對應的黃酮苷(槲皮素、木犀草素、山萘酚、芹菜素、黃芩素、蘆丁、木犀草苷、紫云英苷、野漆樹苷和黃芩苷)(化學結構式如圖1所示),通過鹽酸水解,乙醇溶液提取,HLB固相萃取柱凈化,液相色譜-質譜/質譜法對苷元進行定量測定,從而建立了山銀花中槲皮素、木犀草素、山萘酚、芹菜素和黃芩素5種主要黃酮苷元含量的測定方法。該方法通過對黃酮苷元的檢測,可更為全面地反映出各苷元所代表的黃酮類化合物總量。
1 實驗部分
1.1 儀器與試劑
API 4000型三重四極桿串聯質譜儀(配電噴霧離子源ESI,美國AB公司),1100型液相色譜儀(美國Agilent 公司),Milli-Q Synergy 185超純水器(美國Millipore公司),IKA MS3 Basic型渦旋器(德國IKA公司),24孔固相萃取裝置(美國Supelco公司),Heraeus Multifuge X1R型臺式離心機(美國Thermo 公司),G&G JJ500型電子天平(中國雙杰公司),Mettler AE260(瑞士Mettler Toledo公司)。
甲醇(色譜純,Tedia公司)、甲酸(質譜級,Scharlau公司)、乙腈(色譜純,Scharlau公司)、乙醇(分析純,華東醫藥股份有限公司)、鹽酸(優級醇,永華化學科技(江蘇)有限公司)、叔丁基對苯二酚(TBHQ,分析純,Aladdin公司)、L-(+)-抗壞血酸(分析純,廣東光華科技股份有限公司);CNW C18固相萃取柱(6 mL,500 mg,Anpel公司),Waters C18固相萃取柱(6 mL,500 mg,Waters公司),Waters HLB固相萃取柱(6 mL,200 mg,Waters公司),Biocomma HLB固相萃取柱(6 mL,200 mg,Biocomma公司);色譜柱:Mightysil RP-18(3 μm,4.6 mm×150 mm,日本關東化學株式會社)和 Inertsil ODS-3(3 μm,4.6 mm×150 mm,日本島津公司)。
標準物質:槲皮素(純度96.4%,ChromaDex公司),木犀草素(純度96.9%,ChromaDex公司),山萘酚(純度95.5%,中國食品藥品檢定研究院),芹菜素(純度98.5%,Bepure公司),黃芩素(純度99.2%,ANPEL公司),蘆丁(純度95.0%,TRC公司),木犀草苷(純度98.9%,ANPEL公司),紫云英苷(純度99.1%,ANPEL公司),野漆樹苷(純度98.9%,ANPEL公司),黃芩苷(純度96.8%,ChromaDex公司)。用甲醇將各化合物標準品溶解,稀釋并定容獲得10 mg/mL的標準儲備液,根據需要用甲醇將儲備液稀釋至所需濃度。
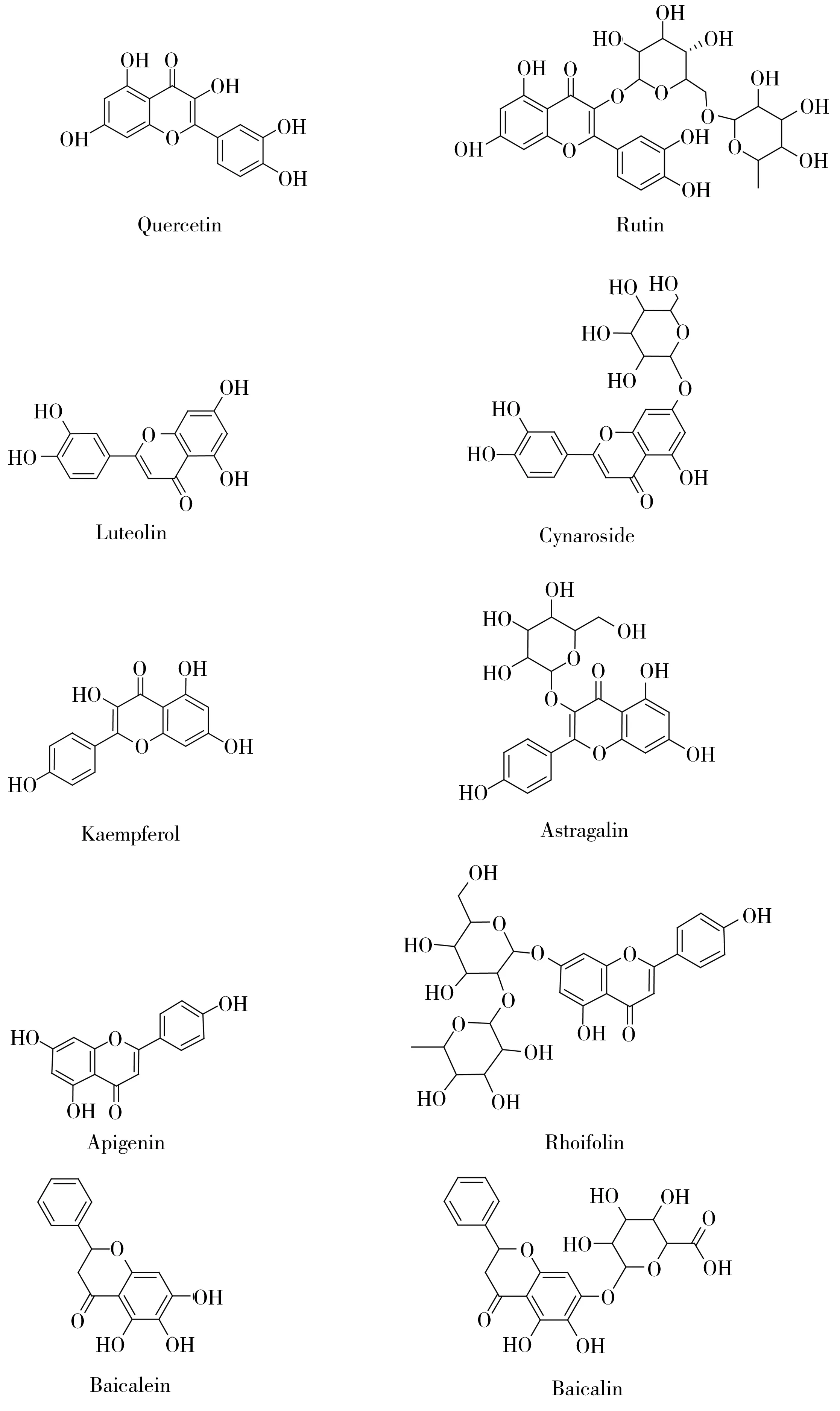
圖1 5種主要黃酮苷元及其對應黃酮苷的化學結構式
1.2 實驗條件
1.2.1 色譜條件色譜柱:Mightysil RP-18(3 μm,4.6 mm×150 mm),流動相:甲醇(A)-0.15%甲酸溶液(B),梯度洗脫程序:0~10.0 min,40%~75%A;10.0~15.0 min,75%~90%A;15.0~22.0 min,90%A;22.0~23.0 min,90%~40%A;23.0~30.0 min,40%A;流速:0.4 mL/min;進樣量:20 μL;柱溫25 ℃。
1.2.2 質譜條件離子源:電噴霧離子源(ESI);掃描模式:負離子掃描;監測方式:多反應監測(MRM);電噴霧電壓(IS):-4 500 V;霧化氣壓力(GS1):289 kPa(42 psi);氣簾氣壓力(GS2):310 kPa(45 psi);輔助氣流速(CUR):172 kPa(25 psi);離子源溫度(TEM):540 ℃;其它質譜參數如表1所示。
1.3 實驗方法
稱取0.5 g(精確至0.01 g)試樣置于150 mL圓底燒瓶中,加0.5 g TBHQ,36 mL 50%的乙醇溶液和4.0 mL濃鹽酸,在氮氣保護下,水浴加熱回流120 min,氮氣保護下冷卻后,加水定容至50 mL,取上清液1 mL,加入9 mL水,混勻并轉移至HLB固相萃取柱中(依次用5 mL甲醇和5 mL 5%甲醇溶液活化),加入5 mL 10%甲醇溶液淋洗,抽干后,加10 mL甲醇進行洗脫,控制流速為1~2 mL/min,收集全部洗脫液,甲醇定容至10 mL,搖勻,過0.22 μm有機濾膜,取1.0 mL定容溶液,加40%甲醇溶液稀釋定容至25 mL,混合均勻,供LC-MS/MS測定。
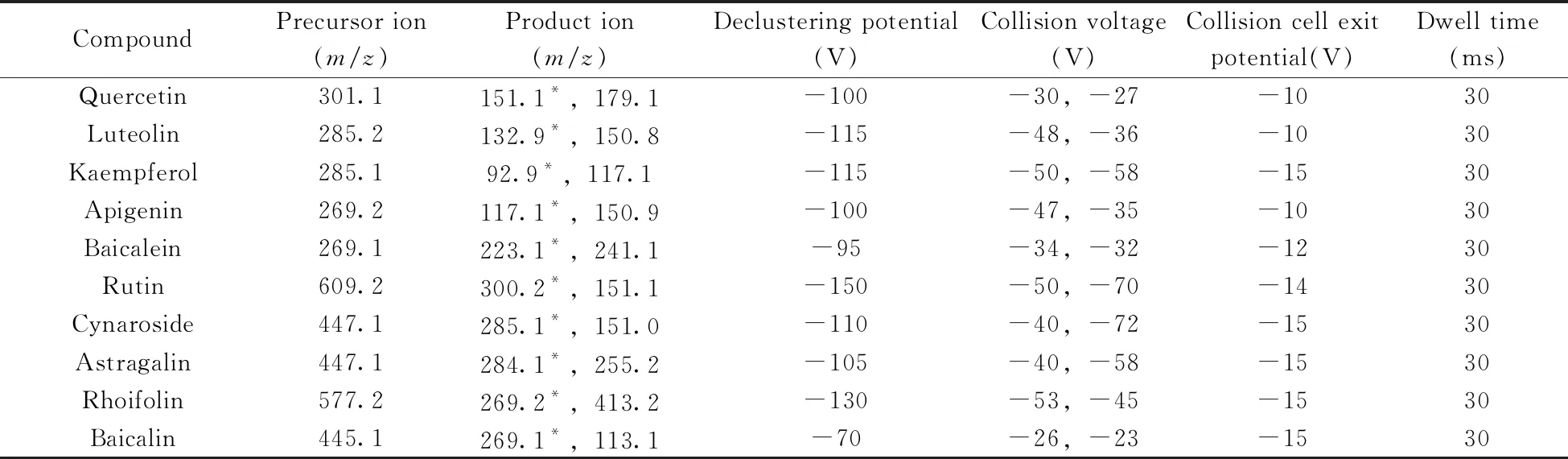
表1 各化合物的基本信息及質譜測定條件
2 結果與討論
2.1 水解與提取條件的選擇
浸提、超聲、回流等技術通常用于中草藥中有效成分的提取,參考中國藥典[1]的方法,實驗首先采用超聲方式進行提取,結果表明超聲環境下黃酮苷幾乎不發生水解反應。
對蘆丁、木犀草苷、紫云英苷、野漆樹苷和黃芩苷在不同含量乙醇溶液(30%、50%、70%),鹽酸(5%、10%和12.5%)條件下,于氮氣保護下水浴加熱回流不同時間(30、60、90、120、180 min)進行實驗,同時對比TBHQ和抗壞血酸兩種不同抗氧化劑的保護情況。結果表明,在無抗氧化劑時,槲皮素、山萘酚和黃岑素的回收率低于50%。在抗壞血酸存在下,槲皮素、山萘酚和黃岑素的回收率無明顯提高,而加入TBHQ后,槲皮素、山萘酚、黃岑素的回收率可達70%以上。乙醇溶液的含量變化對結果無明顯影響,考慮到黃酮苷和苷元的溶解性差異,實驗選擇50%的乙醇進行實驗。在50%乙醇溶液中,當鹽酸濃度低于10%和水解時間小于120 min時,約20%的黃岑苷未水解。實驗最終確定的水解與提取條件為:采用10%鹽酸和50%乙醇溶液,在TBHQ和氮氣保護下水解120 min。在上述條件下對10 ng/mL黃酮苷元進行實驗,發現水解過程中各苷元的損失率均低于20%,有效保障了黃酮苷元的回收率。
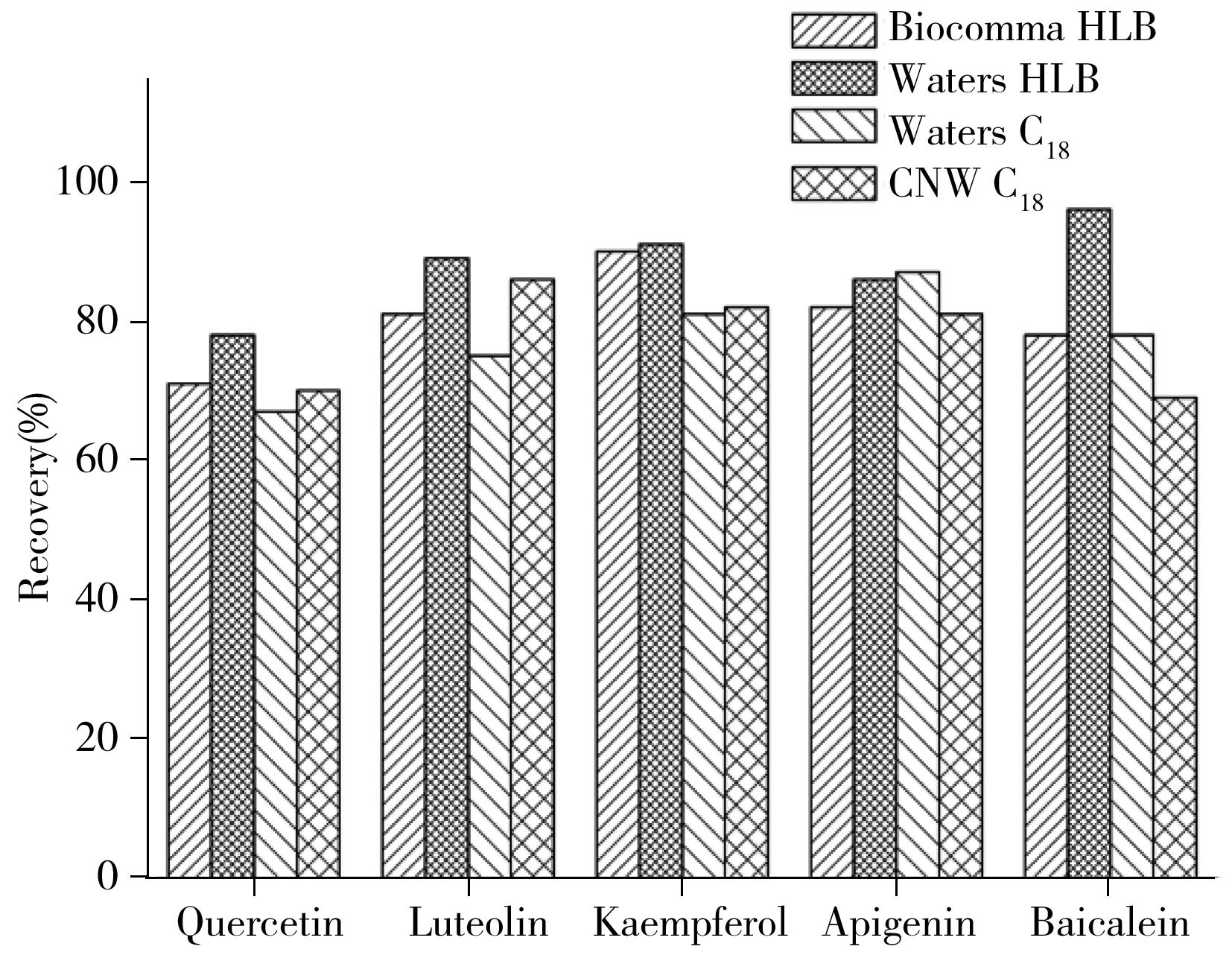
圖2 不同固相萃取柱的凈化情況
2.2 凈化條件的選擇
考察了C18和HLB兩種類型固相萃取凈化柱對水解提取溶液的凈化情況,以降低基質效應對LC-MS/MS定量測定的背景干擾,提高檢測結果的準確性。實驗取1 mL 100 ng/mL的黃酮苷元標準溶液(50%乙醇溶液,含10%鹽酸和0.5 g TBHQ),加入9 mL水,混勻并轉移至各固相萃取柱中,分別采取10 mL的10%、20%、30%、40%、50%甲醇溶液淋洗和甲醇洗脫。結果如圖2所示,50%甲醇溶液淋洗時,C18和HLB固相萃取柱中槲皮素、木犀草素、山萘酚、芹菜素和黃芩素均可以較好地被保留,甲醇洗脫回收率均大于65%,其中槲皮素和黃芩素在C18柱的凈化回收率比HLB柱的凈化回收率低5%~15%。Waters和Biocomma的HLB回收率無明顯差異,但由于黃芪素采用Waters HLB的凈化結果較好,且試驗中Waters HLB的穩定性更好。因此,實驗最終采用Waters HLB固相萃取柱開展方法學考察。
2.3 色譜條件的考察
對比了Mightysil RP-18(3 μm,4.6 mm×150 mm)和 Inertsil ODS-3(3 μm,4.6 mm×150 mm)兩種色譜柱以甲醇或乙腈為有機相,0.15%甲酸溶液或水為水相時對目標化合物槲皮素、木犀草素、山萘酚、芹菜素和黃芩素5種黃酮苷元,及其對應的黃酮苷物質蘆丁、木犀草苷、紫云英苷、野漆樹苷和黃芩苷(質量濃度均為2 ng/mL)的分離情況。
研究表明,相同色譜條件下,黃芩苷在Mightysil RP-18柱中分離時譜峰峰形更好,且信號提升約4倍。在相同色譜柱和梯度洗脫程序下,甲醇為有機相時大部分化合物的響應信號和峰形優于乙腈(其中野漆樹苷、蘆丁、木犀草苷在甲醇為流動相時響應信號提升3~5倍)。在相同色譜柱下,以甲醇為有機相,對比了水和0.15%甲酸溶液為水相的分離情況,以Mightysil RP-18色譜柱為例,在0.15%甲酸溶液中信號的峰形和響應優于水(其中黃芩素提升約5倍)。
因此,實驗最終采用Mightysil RP-18為分離柱,甲醇為有機相,0.15%甲酸溶液為流動相。在該色譜條件下,分子離子峰相同的木犀草素和山萘酚,芹菜素和黃芩素,木犀草苷和紫云英苷可得到很好地分離(如圖3)。
圖3 液相色譜-質譜/質譜法對目標物(2 ng/mL)的分離結果
2.4 質譜條件的優化
對各黃酮苷和苷元的標準溶液分別進行稀釋,采用流動注射的方式在負離子模式下進行母離子全掃描,確定分子離子峰,再分別以待測化合物的分子離子為母離子,對其子離子進行全掃描。按照歐盟EC/657指令和《質譜分析方法通則》的要求,選擇兩個特征子離子對目標化合物進行定量確證,以其中信噪比高、峰形好、干擾小的離子對作為定量離子對。以多反應監測負離子模式優化各種質譜參數,獲得的最佳質譜條件見表1。
2.5 基質效應的考察
通過對比相同濃度的溶劑工作液(稀釋溶劑為流動相初始比例:甲醇-0.15%甲酸溶液,體積比4∶6)和經過前處理的基質加標溶液中各化合物的信號響應強度,計算離子抑制率[離子抑制率(%)=(溶劑工作液中響應強度-基質加標溶液響應強度)×100%/溶劑工作液中響應強度]以考察基質效應情況[23]。如表2所示,實驗條件下,配制質量濃度為4 ng/mL的溶劑工作液和基質加標溶液,得到各化合物定量離子對的離子抑制率均小于15%,即該條件下獲得的基質溶液無明顯基質效應。因此采用甲醇-0.15%甲酸溶液(體積比4∶6)配制工作溶液進行定量計算。
2.6 線性關系、定量下限及回收率
在優化實驗條件下,以待測物標準品的峰面積Y為縱坐標,以其含量X(g/kg)為橫坐標,考察了5種黃酮苷元含量分別為0、0.05、0.1、0.5、1 g/kg(采用40%甲醇溶液配制)時的線性關系。結果顯示,各化合物的線性范圍均為0~1.0 g/kg,相關系數(r2)大于0.995。方法的定量下限(以S/N=10計)為0.005 g/kg(槲皮素),0.01 g/kg(木犀草素和芹菜素)和0.05 g/kg(山萘酚和黃芩素)。
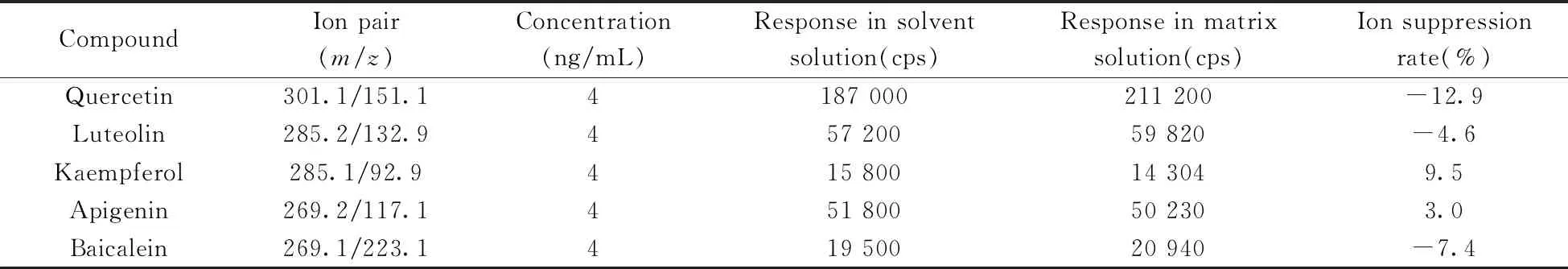
表2 山銀花基質中各化合物的基質效應情況
在優化實驗條件下,對山銀花樣品進行檢測,測定樣品水解后含槲皮素0.17 g/kg、木犀草素0.19 g/kg、山萘酚0.06 g/kg、芹菜素0.05 g/kg和黃芩素0.09 g/kg。對樣品進行3個濃度水平的加標回收實驗,加入各黃酮苷(添加量相當于水解后槲皮素和木犀草素含量分別為0.10、0.20、0.40 g/kg,山萘酚、芹菜素和黃芩素含量分別為0.05、0.10、0.20 g/kg),每個含量水平取6個平行樣,采用外標法定量。結果如表3所示,方法的平均回收率為70.4%~104%,相對標準偏差(RSD)為4.0%~12%。
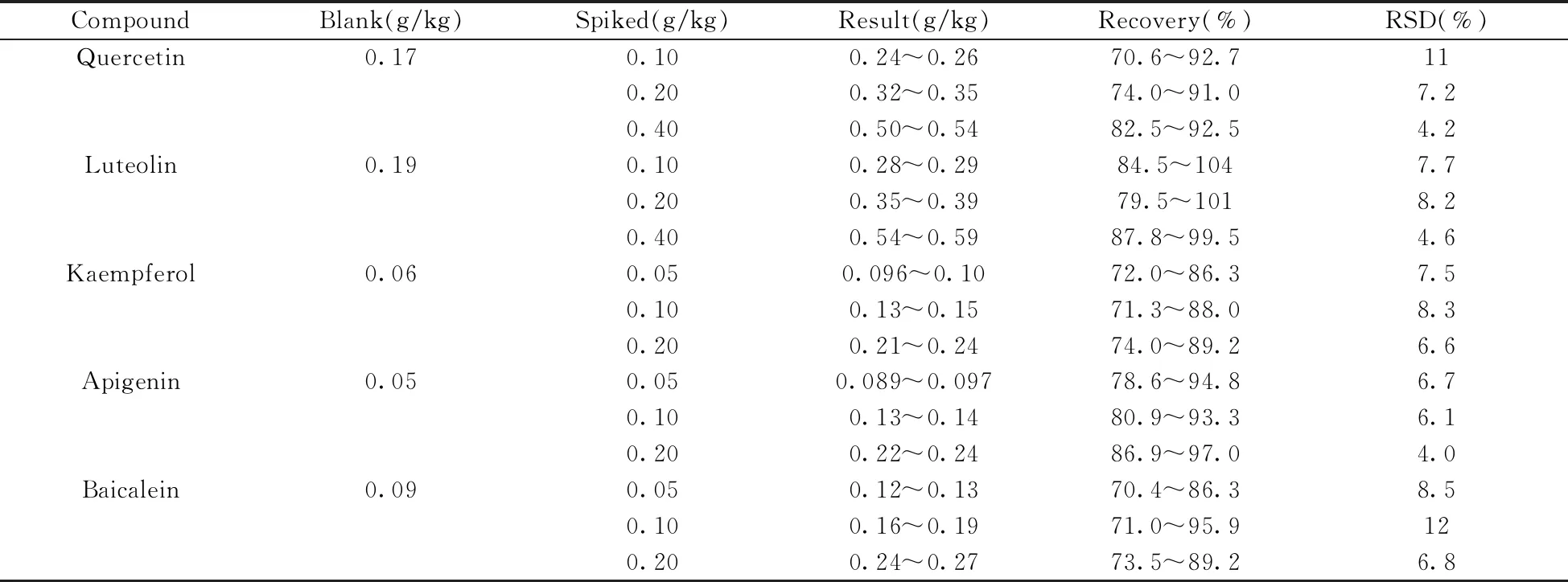
表3 山銀花中5種黃酮苷元在3個加標水平下的回收率及相對標準偏差(n=6)
2.7 實際樣品的檢測
應用本方法對采購的13批山銀花樣品進行測定,結果如表4所示。在山銀花樣品中檢出的5種黃酮苷元中,木犀草素含量最高(0.112~0.453 g/kg),其次是槲皮素(0.030~0.260 g/kg)和芹菜素(0.010~0.050 g/kg),山萘酚和黃岑素的含量最低。
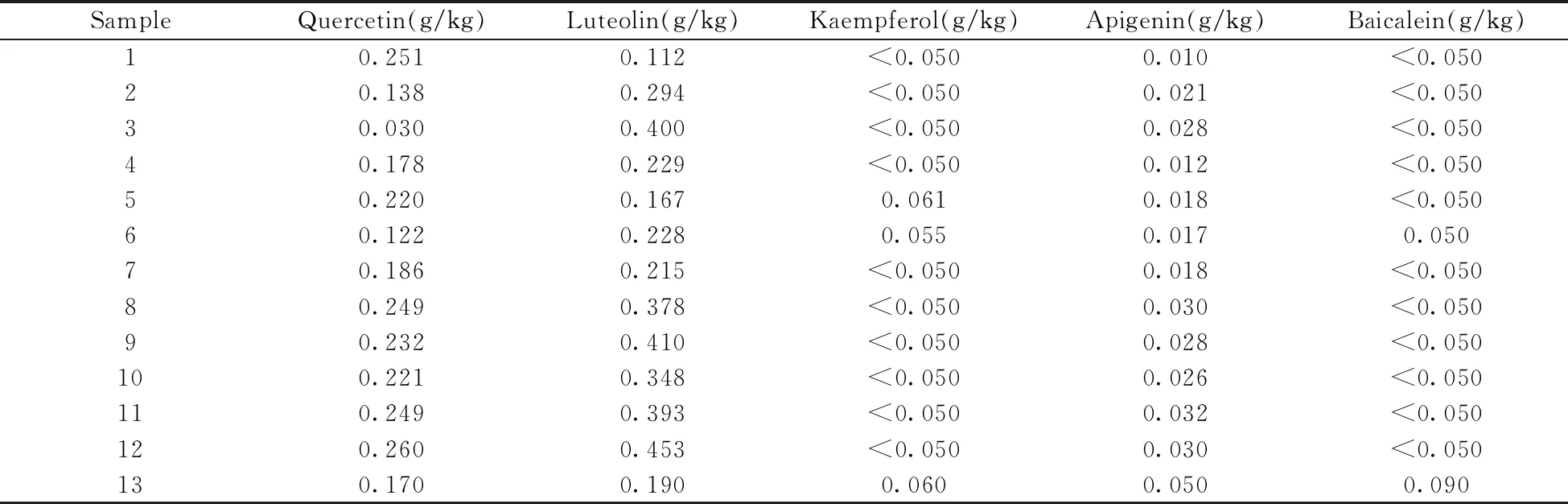
表4 山銀花樣品中的檢測結果
3 結 論
本文通過含有鹽酸的乙醇溶液水解山銀花中5種主要黃酮苷,并對其黃酮苷元進行提取,HLB固相萃取柱凈化,LC-MS/MS法檢測,外標法定量,實現了對山銀花中代表性黃酮苷(蘆丁、木犀草苷、紫云英苷、野漆樹苷和黃芩苷)的有效水解,及水解產物槲皮素、木犀草素、山萘酚、芹菜素和黃芩素含量的測定。該方法通過水解獲得山銀花中的黃酮苷元,并對其含量進行測定,可更準確地獲得山銀花中各主要黃酮類化合物含量的數據,對后續的含量分析以及藥物活性關系和品質研究,具有重要的輔助作用。
猜你喜歡
四川蠶業(2021年2期)2021-03-09 03:15:32
四川蠶業(2021年3期)2021-02-12 02:38:46
中成藥(2018年11期)2018-11-24 02:57:00
中成藥(2017年8期)2017-11-22 03:19:40
中成藥(2017年10期)2017-11-16 00:50:13
中成藥(2017年4期)2017-05-17 06:09:50
哈爾濱醫藥(2016年1期)2017-01-15 13:43:16
天然產物研究與開發(2016年11期)2016-06-15 20:29:17
湖南師范大學自然科學學報(2015年1期)2015-02-27 14:50:04
安徽醫藥(2014年12期)2014-03-20 13:15:15
