3歲男孩反復血小板減少伴溶血性貧血
2021-05-21 07:45:24曾敏慧賀湘玲田鑫黃玉輝
中國當代兒科雜志 2021年5期
曾敏慧 賀湘玲 田鑫 黃玉輝
(1.湖南省人民醫院 / 湖南師范大學附屬第一醫院兒童醫學中心血液腫瘤科,湖南長沙 410005;2.益陽市資陽婦幼保健院兒科,湖南益陽 413001)
1 病例介紹
(1)病史:患兒男,3歲8個月,因反復血小板減少3年余,再發伴發熱2 d入院。患兒生后第1天因氣促1 d、皮膚黃染4 h于外院新生兒科住院,無發熱、血尿、驚厥。查血常規示WBC 18.1×109/L(參考值:5×109/L~12×109/L),Hb 115 g/L(參考值:110~160 g/L),血小板 21×109/L(參考值:100×109/L~400×109/L);肝功能示總膽紅素451.3 μmol/L(參考值:3.4~17.0 μmol/L),間接膽紅素416.3 μmol/L(參考值:3~17 μmol/L);母嬰均為A型Rh陽性血型;考慮診斷為“新生兒高膽紅素血癥、新生兒肺炎”。予美羅培南抗感染、換血治療、輸A型Rh(D)型單采血小板、丙種球蛋白及白蛋白、藍光治療后好轉出院。出院時血小板正常,且出院后半年內定期復查血小板均正常。2年半前患兒因“肺炎”于外院住院,伴皮膚出血點,不伴濃茶色尿,血常規示血小板及Hb均低。考慮血液系統惡性疾病或免疫性血小板減少癥(immune thrombocytopenia,ITP),骨髓穿刺示骨髓增生明顯活躍,粒系增生活躍,形態無異常,紅系增生明顯活躍,淋巴細胞無明顯增減,全片巨核細胞大于200個,血小板成堆分布,不支持惡性血液疾病。治療上予頭孢唑肟抗感染、丙種球蛋白(1 g/kg×1 d)及甲潑尼龍(2 mg/kg×5 d)治療后好轉出院,出院繼續口服潑尼松(每天2 mg/kg,1個月內減停)。2年前再次因發熱、皮膚出血點于外院住院,入院第2~3天出現濃茶色尿,查血小板 15×109/L,Hb 109 g/L, 總 膽 紅 素 56.4 μmol/L, 間 接 膽 紅素47.1 μmol/L,尿潛血陽性,予丙種球蛋白(500 mg/kg×2 d)及潑尼松(每天2 mg/kg,口服4~6周減停)治療,血小板緩慢上升。3個月前患兒因發熱、腹瀉于當地醫院就診,有濃茶色尿,查血小板 18×109/L,Hb 96 g/L,總膽紅素增高,以間接膽紅素增高為主。診斷考慮“血小板減少查因:Evans綜合征、ITP?”,治療上予頭孢他啶抗感染、丙種球蛋白(500 mg/kg×2 d)及甲潑尼龍[10 mg/kg×4 d,后改為潑尼松(每日2 mg/kg ,1個月內減停)]治療后,血小板在 50×109/L~90×109/L間持續3個月后恢復正常。2 d前患兒再次出現反復高熱,最高體溫39.3℃,查血小板60×109/L,無皮膚出血點,無濃茶色尿,無皮膚、鞏膜黃染,為求進一步查明病因收入我科。
(2)個人史、既往史、家族史:患兒系第1胎第1產,既往體質弱,有多次住院病史,生長發育正常,無過敏史。父母身體健康。家族中無近親結婚史,無血尿及血小板減少病史。
(3)入院體檢:體溫39℃,脈搏124次/min,呼吸30次/min,血壓98/52 mm Hg,體重17.7 kg(在-SD至+SD之間),身高106 cm(在-SD至+SD之間)。急性面容,神志清楚,全身皮膚黏膜無黃染、出血點及瘀斑,淺表淋巴結無腫大。心肺體檢無異常,肝、脾肋下未捫及,無四肢、手指畸形。
(4)輔助檢查:血常規(入院當天):血小板 60×109/L(參考值:101×109/L~320×109/L);血常規(入院第5天):血小板 104×109/L,網織紅細胞116×109/L(參考值:24×1012/L~84×1012/L),網織紅細胞比值2.85%(參考值:0.5%~1.5%);肝功能:總膽紅素46.20 μmol/L(參考值:5.1~20.0 μmol/L),間接膽紅素29.58 μmol/L( 參 考 值:5.1~20.0 μmol/L), 乳 酸 脫 氫 酶(lactate dehydrogenase, LDH):570 U/L(參考值:100~240 U/L);免疫全套:IgA 0.41 g/L(參考值:0.58~1 g/L),IgG 6.43 g/L(參考值:6.6~10.39 g/L);尿常規:紅細胞總數 62.2個/μL(參考值:0~18個/μL),尿蛋白 1+(參考值:陰性),尿膽原34 μmol/L(參考值:3.2~16.0 μmol/L)。C 反應蛋白14.84 mg/L(參考值:0~10 mg/L);降鈣素原0.09 ng/mL(參考值:0~0.05 ng/mL);電解質、血糖、凝血功能、腎功能正常, 直接抗人球蛋白試驗(direct antiglobulin test, DAT)、抗核抗體譜、抗中性粒細胞胞漿抗體、抗磷脂抗體譜均陰性。葡萄糖‐6‐磷酸脫氫酶(比值法):1.66(參考值:1.0~1.66);地中海貧血基因:基因型αα/αα,βN/βN;血涂片:成熟紅細胞大致正常,血小板散在分布,未見異型淋巴細胞。
2 診斷思維
患兒病例特點:(1)學齡前期男孩,出生時即起病,新生兒時期有黃疸及換血治療史;(2)主要表現為反復感染伴血小板減少及溶血性貧血;(3)無肝脾、淋巴結腫大;(4)腎功能正常,DAT陰性,總膽紅素升高,以間接膽紅素升高為主,網織紅細胞及LDH增高;(5)骨髓細胞學檢查未見異常。
該患兒臨床表現主要為反復血小板減少伴不同程度的溶血性貧血,診斷從以下兩方面考慮:(1)血小板生成減少:可由于巨核細胞發育不全、受抑或骨髓浸潤所致,臨床常見于急性白血病、骨髓衰竭綜合征、葉酸及維生素B12缺乏、惡性腫瘤骨髓轉移及遺傳性血小板減少癥等,該患兒病程長達3年余,血小板減少可通過抗感染、丙種球蛋白、糖皮質激素治療后緩慢恢復,但患兒網織紅細胞增高,骨髓細胞學檢查提示骨髓增生明顯活躍,未見原始幼稚細胞,未見“老漿幼核”現象,巨核細胞數目無減少,血片未見異常血小板團塊,凝血功能正常,故不支持血小板生成減少相關疾病。(2)血小板破壞增加:分為免疫性血小板減少和非免疫性血小板減少。免疫性血小板減少主要考慮①ITP:該患兒反復血小板減少3年余,既往經抗感染、丙種球蛋白及糖皮質激素治療后血小板能緩慢恢復,但3個月前上述治療無效,且病程中伴有不同程度的溶血性貧血,故不支持慢性ITP。②Evans綜合征:一種罕見的自身免疫性血液系統疾病,為血細胞特異性自身抗體引起的血小板和紅細胞破壞增加[1],臨床上可同時或相繼發生自身免疫性溶血性貧血和ITP,可伴有免疫性WBC減少,好發于兒童,DAT陽性[2‐3],該患兒有反復血小板減少伴溶血性貧血,但DAT陰性,故不支持Evans綜合征。③抗磷脂綜合征:是自身免疫介導的反復動靜脈血栓形成導致的器官缺血或衰竭的一組臨床綜合征,可表現為抗磷脂抗體相關腎病、血小板減少、溶血性貧血等[4],該患兒有血小板減少及溶血性貧血表現,但腎功能正常,抗核抗體譜及抗磷脂抗體陰性,故不支持抗磷脂綜合征。
非免疫性血小板減少多為血小板消耗增多,可見于巨大血管瘤、脈管畸形、脾功能亢進、青紫型心臟病等,但該患兒缺乏相關病史及臨床表現,故不支持。故主要考慮①彌漫性血管內凝血:是許多疾病致病因素導致凝血活化,全身微血管血栓形成、凝血因子大量消耗并繼發纖溶亢進,引起以出血及微循環衰竭為特征的臨床綜合征[5],臨床表現主要為出血、休克或微循環衰竭、微血管栓塞及微血管病性溶血,該患兒有血小板減少、皮膚出血及溶血表現,但缺乏起病的始動因素,無明顯大出血、休克、微循環衰竭的表現,且凝血功能正常,故不支持該病。②溶血尿毒綜合征(hemolytic urothelial syndrome, HUS):是血栓性微血管病(thrombotic microangiopathy, TMA)的常見類型,90%見于兒童,根據發病機制不同,分為典型HUS和非典型HUS。典型HUS為腹瀉相關HUS[6];非典型HUS特指補體替代途徑調控異常引起的血管內皮功能失調和血管微血栓的形成,二者臨床表現均為微血管性溶血性貧血、血小板減少及急性腎損傷[7]。該患兒有反復血小板減少伴溶血性貧血,但無血便,且腎功能正常,不支持HUS。③血栓性血小板減少性紫癜(thrombotic thrombocytopenic purpura, TTP):既往的經典五聯征有發熱、血小板減少、微血管性溶血性貧血、神經系統癥狀及腎功能不全,該患兒有發熱、血小板減少及溶血性貧血病史,且DAT陰性,凝血功能正常,LDH升高,故需考慮該病。若該患兒存在腎功能不全,那么需與非典型HUS鑒別[8‐9]。需檢測TTP特異性指標—血管性血友病因子(von Willebrand factor, vWF) 裂 解 酶(von Willebrand factor‐cleaving protease, ADAMTS13)活性及其抑制性抗體來協助診斷[8],必要時完善基因檢測以確診。
3 進一步檢查
ADAMTS13對診斷TTP具有很高敏感性和特異性,能夠將TTP與其他TMA綜合征(典型HUS、非典型HUS)和血液學其他血細胞減少癥區分開來[10]。ADAMTS13活性:0.23%(參考值:42.16%~126.37%),ADAMTS13活性抑制性抗體陰性(參考值:陰性)。征得患兒家屬同意后,取患兒及其父母外周血各3 mL,提取DNA,進行全外顯子組測序,結果提示ADAMTS13基因存在復合雜合變異:c.1045C>T(p.R349C)和c.1928T>G(p.I643S),前者來源于母親,后者來源于父親,見圖1。根據美國醫學遺傳學與基因組學學會變異分 類 指 南[11],c.1045C>T(p.R349C)在 OMIM 數據庫(https://omim.org)已有該位點關聯先天性血栓性血小板減少性紫癜(congenital thrombotic thrombocytopenic purpura, cTTP)的致病性報道,在千人數據庫(https://www.ncbi.nlm.nih.gov/variation/tools/1000genomes/)正常對照人群中該變異頻率<0.001,符合PM2;已有的功能研究表明該變異為有害作用,符合PS3;多個生物信息學軟件預測為有害,符合PP3;患兒表型具有高度基因特異性,符合PP4;故該變異判定為“可能致病”。c.1928T>G(p.I643S)在千人數據庫正常對照人群中沒有報道,符合PM2;在隱性疾病中反式檢測到致病變異,符合PM3;多個生物信息學軟件預測為有害,符合PP3;患兒表型具有高度基因特異性,符合PP4;故該變異判定為“可能致病”。
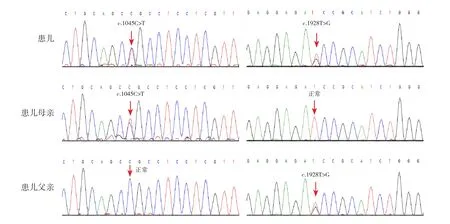
圖1 患兒及其父母ADAMTS13基因Sanger測序圖 患兒ADAMTS13基因存在c.1045C>T和c.1928T>G復合雜合突變,前者來源于母親,后者來源于父親。突變位點如箭頭所示。
4 診斷及依據
診斷:cTTP。依據:(1)出生起病,有新生兒黃疸及換血治療史;(2)反復感染后血小板減少伴溶血性貧血;(3)DAT陰性,凝血功能正常,膽紅素及LDH升高;(4)ADAMTS13活性極度缺乏,ADAMTS13活性抑制性抗體陰性,ADAMTS13基因存在復合雜合變異[12]。
5 臨床經過
入院后予頭孢哌酮舒巴坦抗感染及輸注丙種球蛋白后血小板無上升,待ADAMTS13活性檢測提示極度缺乏且ADAMTS13抑制性抗體檢測陰性時考慮cTTP,予輸注新鮮冰凍血漿200 mL后血小板明顯上升,2 d后升至正常,后基因檢測結果支持cTTP。
患兒出院后2個月有發熱等感染癥狀,查血小板>50×109/L,抗感染治療5 d后血小板緩慢升至正常,未予輸注血漿。出院后4個月再次出現發熱,血小板降至29×109/L,間接膽紅素增高,予輸注新鮮冰凍血漿200 mL,2 d后復查血小板恢復正常,膽紅素數天后緩慢降至正常。隨訪至出院后5個月,患兒生存狀態同正常同齡兒。
6 討論
1924年,Moschcowitz[13]首次在臨床上將TTP描述為罕見的致命性TMA。全球TTP發病率約為(1.5~4)/106,以獲得性多見,先天性占所有病例不到5%,兒童TTP發病率約為1/106(其中cTTP占所有兒童病例的1/3)[10,14]。其特征是微血管病性溶血性貧血、嚴重的血小板減少癥伴或不伴彌散的微血管性血小板富集血栓形成導致的器官缺血。cTTP是由于ADAMTS13基因突變所致,該基因位于9q34上,編碼的蛋白是金屬蛋白酶家族的第13個成員,主要在肝星狀細胞中表達,特異性切割具有促血栓作用的大分子vWF多聚體[15‐16]。全世界約有150種ADAMTS13基因突變,以常染色體隱性方式遺傳,70%為錯義突變[17‐18]。
cTTP發病機制為患者在遭受感染、創傷、懷孕等誘因刺激下,內皮細胞產生過多超大型vWF,由于ADAMTS13的缺乏,導致超大型vWF不能被切割成正常生理狀態下的vWF多聚體,超大型vWF進入微循環與血小板異常聚集形成微血栓,致微血管狹窄,造成組織或器官缺血[19]。因此對cTTP患者在未發生嚴重危及生命的大出血時,應避免輸注血小板,防止加重組織器官缺血。當紅細胞通過微血管時,致紅細胞機械性損傷,因此血涂片可見破碎紅細胞,但有10%的患者可能出現陰性結果[10,19],需多次涂片驗證。對于既往用于診斷TTP的五聯征的歷史性臨床研究已經過時,因為這5種癥狀僅在不到10%的急性TTP患者中同時出現[9]。因此,目前臨床工作中,將ADAMTS13 作為TTP的特異性標記,能夠將TTP與其他TMA和其他血細胞減少癥區分開來。
cTTP起病通常發生在嬰兒期(包括新生兒期)或兒童早期(10歲以前),而很少發生在青春期[9]。獲得性TTP在兒童或青少年中報道較少,成人多見,通常起病急劇,病死率高,多需在重癥監護室搶救治療[20]。而cTTP臨床表現的嚴重程度、器官損傷和發作頻率不一致,可能與ADAMTS13突變類型及其他遺傳或環境因素有關[14]。該例患兒3年間反復多次發作,但均無大出血、嚴重的溶血性貧血及神經系統異常(如昏迷等),考慮可能與其基因突變類型有關。
目前,對cTTP的治療仍然主要為補充ADAMTS13,通過應用新鮮冰凍血漿以提高ADAMTS13活性[10,21]。臨床中對緩解期的cTTP患者,建議輸注血漿或采取臨床觀察;對合并有臟器缺血損害的嚴重發作病例或慢性反復發作病例推薦預防治療:每2~4周輸注1次新鮮冰凍血漿以維持血小板在安全水平且臨床無發作;對無臟器受累患兒給予按需治療:當血小板<50×109/L時給予血漿;對于無癥狀的cTTP,強烈建議孕婦在懷孕期間進行預防性血漿輸注[22]。雖然有指南提出每2~4周以血漿10~15 mL/kg輸注可有效預防急性發作,但實際工作中,由于患者反復來醫院輸注血制品給患者家庭帶來負擔,降低其生活質量,同時有感染傳染病風險及相關不良反應,且該患兒3年來急性發作時并無嚴重的大出血、重度貧血或明顯的神經系統癥狀及腎功能不全等表現,因此我們綜合考慮該患兒采取按需輸注原則。隨著對TTP的研究進展[23],在治療方面也給cTTP患者帶來了新的希望。BAX930是一種由中國倉鼠卵巢基因工程細胞系合成的完全糖基化的重組人ADAMTS13 蛋白[24‐25],2017 年已完成 I期多中心臨床研究,結果顯示重組ADAMTS13安全有效,患者對藥物耐受性良好,免疫原性試驗均陰性[26]。2018年1月起開展來自10個國家的多中心Ⅲ期臨床試驗(NCT03393975),目前正在招募中,預計在2023年完成,以評估BAX930在預防和按需治療ADAMTS13活性<10%的重度 cTTP 患者中的安全性和有效性。這項Ⅲ 期臨床試驗的完成對于cTTP的患者來說將會獲益[27]。
7 結語
cTTP臨床極其罕見,由ADAMTS13基因突變導致ADAMTS13活性降低引起的微血管病性溶血性貧血,嚴重的血小板減少伴或不伴彌散的微血管性血小板富集血栓形成導致的器官缺血。主要表現為嬰幼兒期的黃疸或需換血治療,通常為感染后發生的血小板減少伴溶血性貧血,可有腎功能損害、神經系統癥狀及發熱;實驗室檢查為DAT陰性、凝血功能正常,膽紅素、網織紅細胞及LDH升高,血涂片見破碎紅細胞等,對于這些患者建議完善ADAMTS13活性檢測,避免漏診[8]。臨床工作中,尤其對于小嬰兒存在黃疸且伴有不明原因的血小板減少時,我們需要關注cTTP[8],可完善ADAMTS13活性及抑制性抗體檢測,對ADAMTS13活性極度缺乏及抑制性抗體陰性時,可予血漿輸注,同時完善基因檢測以盡早明確診斷。
