酯交換-熔融縮聚法合成PEF工藝及性能研究
2021-06-09 03:15:32王樹霞戴鈞明黃洛瑋王玉合
合成技術及應用 2021年1期
關鍵詞:催化劑
王樹霞,戴鈞明,黃洛瑋,王玉合,司 虎
(1. 中國石化儀征化纖有限責任公司研究院,江蘇儀征 211900; 2. 江蘇省高性能纖維重點實驗室,江蘇儀征 211900)
隨著石油資源日益短缺及可持續發展理念的深入,生物法合成化工原料逐漸成為趨勢。現在常見的生物基高分子聚乳酸(PLA)、聚丁二酸丁二醇酯(PBS)、聚羥基脂肪酸酯(PHAs)等存在共同的缺點:熔點低、力學性能不足、耐熱性差,其主要原因是脂肪鏈分子結構,剛性差。來源于纖維素、果糖或海洋大型藻類等生物基的2,5-呋喃二甲酸(FDCA)結構中含有剛性呋喃環,所制得的PEF具有優異的熱學-力學性能以及比聚對苯二甲酸乙二醇酯(PET)高一個數量級的氣體阻隔性,在纖維、薄膜以及包裝材料等領域有望替代PET[1],發展潛力巨大。
合成PEF主要有溶液縮聚法、酯化-熔融縮聚法、酯交換-熔融縮聚法、固相縮聚法和開環聚合法,其中研究最多的是酯化-熔融縮聚法。由于DMFD相比FDCA更易提純,且可避免脫羧反應[2]。本研究以DMFD和乙二醇為原料,合成PEF,考察酯交換催化劑用量、酯交換溫度、縮聚溫度對反應速率的影響,并對合成的產品進行了結構性能表征,為PEF工業化生產和應用提供數據支撐。
1 試 驗
1.1 原料
2,5-呋喃二甲酸二甲酯,工業級,中科院寧波材料所;乙二醇,工業級,中國石化揚子石油化工有限公司;酯交換催化劑,化學純,南京化學試劑廠;乙二醇銻,工業級,江蘇大康實業公司。
1.2 設備
聚合反應釜,2.0L,自制,熱媒加熱,錨式攪拌;激光粒度儀,Marstersizer2000型,英國馬爾文公司;掃描電鏡,Nova Namo SEM450型,美國FEI公司;核磁共振儀,AV-600型,德國Bruker公司;相對黏度儀,Viscotek Y501型,英國馬爾文公司;傅里葉變換紅外光譜儀,IS10型,美國賽默飛世爾公司;熱分析儀,DSC 7型,美國Perkin-Elmer公司;熱重分析儀,Diamond型,美國Perkin-Elmer公司。
1.3 試驗過程
在2 L聚合反應釜中加入一定量的DMFD、EG、酯交換催化劑和乙二醇銻催化劑,進行酯交換,酯交換反應結束后,進行縮聚反應,攪拌器電流達到一定值時,氮壓出料,冷卻切粒,制得PEF聚酯。PEF的酯交換-熔融縮聚反應過程如圖1所示,主要工藝如表1所示。

表1 試驗主要工藝
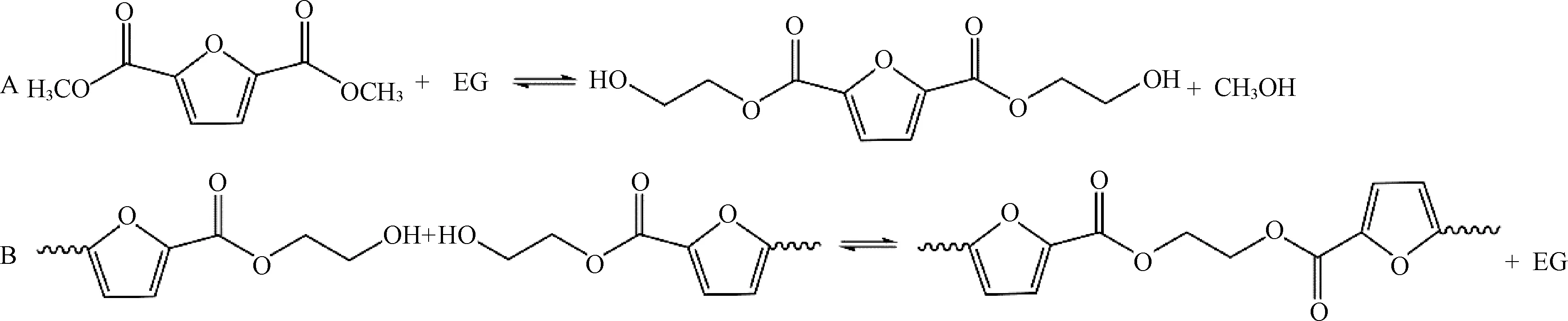
圖1 酯交換-熔融縮聚法制PEF:(A)酯交換階段;(B)熔融縮聚階段
1.4 分析測試
1.4.1 粒徑分布測試
用馬爾文激光粒度儀干法測試DMFD樣品,振動進樣速度50%,分散氣壓0.1 MPa。
1.4.2 掃描電鏡測試
將DMFD樣品粘在導電膠上噴金,放大倍率500倍。
1.4.3 特性黏度測試
溫度(25±0.1)℃,溶劑為苯酚-四氯乙烷(質量比3∶2)。
1.4.4 紅外光譜測試
將樣品熔融熱壓,在4 000~400 cm-1范圍內攝譜。
1.4.5 核磁測試
以二甲基亞砜DMSO為溶劑,四甲基硅烷(TMS)為內標,分析PEF的分子結構。
1.4.6 熱性能測試
在氮氣保護下,以10 ℃/min的升溫速率從25 ℃升高到290 ℃,保持5 min,然后以400 ℃/min的速率降至25 ℃,再以10 ℃/min的升溫速率從25 ℃升高到290 ℃,保持5 min,最后以10 ℃/min的速率降至100 ℃。
1.4.7 熱失重測試
空氣氛圍,通氣量20 mL/min,升溫速率為10 ℃/min,由室溫升至650 ℃。
2 結果與討論
2.1 DMFD形貌及結構
DMFD的粒徑分布如圖2所示,2,5-DMFD的掃描電鏡觀察結果如圖3所示。
由圖2和圖3可以看出,DMFD粉體中值粒徑42.8 μm,且分布較寬,外觀形貌近似針狀。2,5-DMFD中值粒徑較小且為針狀形貌,因此其比表面積較大,增加了酯交換反應時與EG的非均相接觸面積,利于酯交換反應。根據PTA/EG漿料配制經驗[3],2,5-DMFD粒徑分布寬,配成的EG/2,5-DMFD漿料穩定性好,利于酯交換反應。

圖2 DMFD粒徑分布

圖3 DMFD的SEM圖像
2.2 合成工藝影響研究
2.2.1 酯交換溫度影響
在添加相同酯交換催化劑條件下,酯交換反應溫度對甲醇餾出速度的影響如圖4和5所示。

圖4 酯交換反應釜液溫曲線
酯交換反應溫度升高,甲醇餾出速率加快,酯交換速度明顯變快。酯交換溫度為160~210 ℃時,酯交換反應速率隨升溫速率增加而加快。這是因為酯交換反應為吸熱反應,提高反應溫度有利于向正反應方向進行。甲醇餾出量與酯交換反應時間呈較好的線性關系,符合一級反應特征。

圖5 甲醇餾出速度擬合曲線
2.2.2 酯交換催化劑添加量影響
相似反應溫度條件下,催化劑添加量對酯交換速度的影響如圖6和圖7所示。

圖6 不同催化劑用量下甲醇餾出情況

圖7 催化劑用量對甲醇餾出速率影響
當酯交換催化劑添加量從300 μg/g逐步增加至500 μg/g時,甲醇餾出速度逐漸加快,當酯交換催化劑添加量由從500 μg/g 增加至600 μg/g時,甲醇餾出速率基本不變。此趨勢與酯交換法合成PET相似[4]。酯交換反應為配位催化反應,首先催化劑與乙二醇形成醇化物,然后由醇化物上的金屬提供空軌道與呋喃二甲酸二甲酯的羰基上氧配位,使羰基極化而使羰基上碳原子的正電性增大,進而使醇化物上的乙二醇基團與羰基碳結合。因此在一定范圍內,當催化劑含量增加,加速酯交換反應速率。
2.2.3 縮聚溫度影響
縮聚溫度對縮聚反應的影響如圖8和圖9所示,縮聚溫度由240 ℃升至245 ℃,隨著反應時間的增加PEF特性黏度增加幅度相當,當縮聚溫度由245 ℃升至255 ℃,PEF特性黏度隨縮聚溫度升高增長較快,即PEF分子量呈快速增長趨勢。255 ℃聚合170 min得到滿足基本使用要求的特性黏度為0.601 dL/g的聚合物PEF,說明在一定范圍內,提高縮聚溫度,有利于分子鏈快速增長。縮聚反應是可逆平衡化學反應,既有鏈增長反應,也有大分子鏈的熱降解反應和鏈端熱降解反應,同時還有擴散過程的影響。呋喃環為不對稱芳香環,鏈段不易翻轉,此外呋喃環存在永久偶極矩,增大了鏈間相互作用力,鏈段運動能力較弱[5],低溫時反應能力較低。

圖8 縮聚溫度對縮聚反應的影響
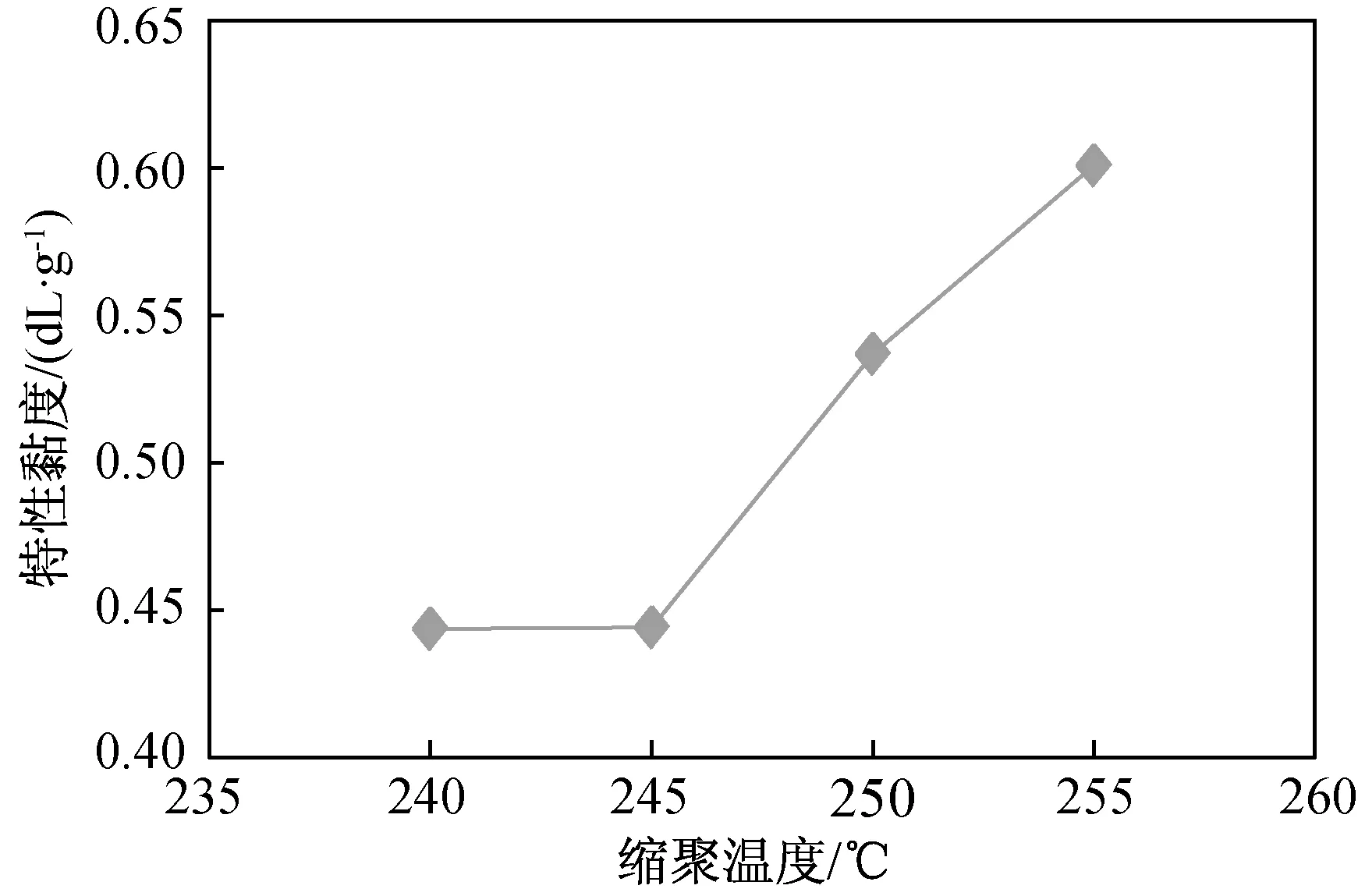
圖9 縮聚溫度對PEF特性黏度的影響
2.3 PEF結構與性能
2.3.1 紅外分析


圖10 PEF的紅外譜圖
2.3.2 核磁分析
酯交換熔融縮聚法制得的PEF的1H-NMR如圖11所示。由圖11可知,7.15處為呋喃環上的質子峰;4.57處為—OCH2—CH2—上的質子峰;4.46和3.95處分別為與端羥基相連亞甲基上的質子峰。結合FTIR和1H-NMR對產物結構的分析表明利用酯交換熔融縮聚法制備了PEF[8-10],并且合成過程中存在著較少的降解副反應。

圖11 PEF的1H-NMR譜圖
2.3.3 熱性能研究
酯交換熔融縮聚法制得的PEF的熱分析測試結果如圖12和表2所示。

(a) PEF的DSC曲線

表2 PEF的DSC與TGA測試結果
由圖12和表2可知,第一條升溫線Tg為80.78 ℃,沒有冷結晶峰,熔融峰值為212.0 ℃,熔融峰的焓值只有0.96 J/g,消除熱歷史后的第二條升溫線Tg為88.97 ℃,起始熱分解溫度為396.52 ℃,比李連貴[11]報道結果略高,酯交換-熔融縮聚法制得的PEF熱穩定性較好,但比PET熱穩定性略差[8,10-11]。因為呋喃環是含氧五元雜環,其電負性比苯環大,在高分子鏈段中由于呋喃環的存在會形成分子間氫鍵,呋喃環中的原子個數小于苯環,其穩定性比苯環差。
3 結 論
通過酯交換熔融縮聚法合成了PEF,研究了催化劑用量、酯交換反應溫度及縮聚反應溫度對反應速率的影響,對合成的PEF結構性能進行了表征。
a) DMFD與EG酯交換反應動力學符合一級反應特征,當酯交換催化劑添加量從300 μg/g增加至500 μg/g時,甲醇餾出速度隨催化劑添加量增加而增長,酯交換反應速率隨催化劑添加量增加而呈線性增長;當添加量超過500 μg/g時,反應速率不再增加;酯交換溫度為160~210 ℃時,酯交換反應速率隨升溫速率增加而加快。
b) 當縮聚溫度由245 ℃升至255 ℃,PEF分子量隨縮聚溫度升高呈快速增長趨勢。
c) 酯交換-熔融縮聚法制得PEF的玻璃化轉變溫度為88.97 ℃,起始熱分解溫度為396.52 ℃,熱穩定性較好。
猜你喜歡
大自然探索(2023年7期)2023-11-14 13:08:06
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
智富時代(2018年3期)2018-06-11 16:10:44
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
超硬材料工程(2016年1期)2016-02-28 22:20:04
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
合成化學(2015年4期)2016-01-17 09:01:27
應用化工(2014年3期)2014-08-16 13:23:50
