我國藥品緊急使用的制度問題及對策建議*
2021-08-05 00:28:36徐東紫歐陽昭連
中國藥業 2021年14期
陳 娟,盧 巖,徐東紫,張 婷,嚴 舒,歐陽昭連
(中國醫學科學院醫學信息研究所,北京 100020)
進入21世紀以來,全球遭遇了多次突發公共衛生事件,包括重癥急性呼吸綜合征(SARS)、中東呼吸綜合征(MERS)、埃博拉病毒、寨卡病毒及新型冠狀病毒肺炎(簡稱新冠肺炎)等疫情,這些事件給醫療系統和社會經濟造成了巨大沖擊,如何有效應對突發公共衛生事件是各國面臨的重要議題[1]。一種新的突發公共衛生事件發生時,短時間內常缺乏經充分證實有效的可用藥品,而研發一種新的藥品需要較長時間,這就意味著必須選擇一些已上市藥品或在研藥品緊急使用[2-3]。然而,緊急使用未上市藥品和超藥品說明書使用已上市藥品在我國缺乏相應法律基礎[4-5]。我國針對突發公共衛生事件建立的藥品特別審批制度及當前已有的其他特殊審批通道均無法解決應急狀態下的藥品緊急使用問題[6-7]。美國針對突發公共衛生事件建立了藥品緊急使用授權制度,且該制度經過十幾年的不斷完善已趨于成熟[8-9]。本研究中擬采用文獻調研法和專家咨詢法探討我國新冠肺炎疫情期間使用未上市藥品或超藥品說明書用藥的制度問題、我國現有特別審批制度及其他特殊審批通道在應對突發公共衛生事件中的不足,并借鑒美國藥品緊急使用授權制度,為我國藥品緊急使用授權制度的建立提供建議。現報道如下。
1 我國藥品緊急使用的制度缺陷
1.1 新冠肺炎疫情涉及藥物
治療性藥物:新冠肺炎疫情暴發以來,國家衛生健康委員會先后發布了多個版本的診療方案,其中推薦了多種治療藥物,包括未上市藥品(如瑞德西韋),適應證或用法用量超藥品說明書的已上市藥品(如α-干擾素、洛匹那韋利托那韋、利巴韋林、氯喹、羥氯喹、阿比多爾、法匹拉韋、達蘆那韋考比司他、奧司他韋、托珠單抗、烏司他丁、甘草酸制劑等),在患者救治中發揮了重要作用[10-12]。但其藥品說明書中大多無新冠肺炎適應證,部分藥品尚未上市[3,13]。
預防性疫苗:2020年6月29日,康希諾生物股份有限公司宣布,其與軍事科學院軍事醫學研究院生物工程研究所聯合開發的腺病毒載體疫苗(Ad5-nCoV)已于2020年6月25日獲得中央軍委后勤保障部衛生局頒發的軍隊特需藥品批件,獲準僅限于軍隊內部使用,當時該疫苗尚處于臨床試驗階段,并未正式獲批上市。2020年8月22日,國家衛生健康委科技發展中心主任、國務院聯防聯控機制科研攻關組疫苗研發專班工作組組長鄭忠偉在央視《對話》欄目上介紹,我國已于2020年7月22日正式啟動國藥集團2款滅活疫苗的緊急使用,截至8月底已在醫務人員、防疫人員、邊檢人員等特殊人群中接種了數十萬人次,當時該疫苗同樣處于臨床試驗階段,并未正式獲批上市。
1.2 相關法律法規
突發公共衛生事件應急狀態下的藥品使用應遵循的上位法,包括突發公共衛生事件應急相關法律法規和藥品注冊審評相關法律法規[14-21]。現對兩類法律法規文件中與“藥品應急”有關的條款進行如下梳理(見圖1)。

圖1 我國在新冠肺炎疫情期間使用未上市藥品或超藥品說明書用藥應遵循的法律依據Fig.1 Legal terms that should be followed when using investigational drugs or off-label drugs during the COVID-19 epidemics in China
突發公共衛生事件應急相關法律法規:我國應對突發公共衛生事件的法律依據主要包括《中華人民共和國傳染病防治法》(簡稱《傳染病防治法》)、《中華人民共和國突發事件應對法》(簡稱《突發事件應對法》)和《突發公共衛生事件應急條例》[16-18]。《傳染病防治法》于1989年2月21日通過,之后分別于2004年和2013年進行了修訂,是我國應對傳染病的主要法律依據。最新版中與藥品應急有關的條款包括第四十九條和第六十三條,前者對傳染病暴發時的藥品生產和供應作了相應描述,后者對防治傳染病的藥品儲備問題作了責任說明。《突發事件應對法》于2007年8月30日通過,自2007年11月1日起開始實施,該法律的頒布標志著我國突發事件應對正式進入法制化軌道。其中第三十二條對應急所需物資的監管、生產、儲備、調撥和配送的權責分配作了相應說明。《突發公共衛生事件應急條例》于2003年5月9日頒布,成為我國應對突發公共衛生事件的主要依據,該條例于2011年進行了修訂,最新版中與藥品應急有關的條款包括第十六條、第十七條和第三十二條,上述條款對應急所需藥品的儲備、生產和供應問題作了說明(見表1)。然而,上述文件均未明確提及應急狀態下的藥品緊急使用問題。
藥品注冊審評相關法律法規:我國藥品注冊審評相關法律依據主要包括《中華人民共和國藥品管理法》(簡稱《藥品管理法》)、《中華人民共和國疫苗管理法》(簡稱《疫苗管理法》)、《藥品管理法實施條例》和《藥品注冊管理辦法》[14,15,20]。我國《藥品管理法》于1984年通過,于2001年和2019年進行了2次修訂,最新版《藥品管理法》從2019年12月1日起開始實施,其中第二十三條、第二十六條和第九十六條分別對拓展性臨床試驗制度、附條件批準程序和優先審評審批程序進行了闡述,第九十二條對藥品儲備和緊急調用作了相關說明。我國《疫苗管理法》于2019年12月1日開始實施,其中第二十條指出,重大突發公共衛生事件急需的疫苗可以附條件批準,特別重大突發公共衛生事件所需疫苗可以在一定時間和范圍內緊急使用;第二十八條規定,應對突發事件急需的疫苗可免于批簽發。我國現行《藥品管理法實施條例》是2002年根據舊版《藥品管理法》頒布的,其中未提及藥品緊急使用相關內容,新版《藥品管理法實施條例》尚在制訂中。我國《藥品注冊管理辦法》于2002年通過,分別于2005年、2007年和2020年進行了修訂,最新版《藥品注冊管理辦法》于2020年7月1日起開始實施,其中第五十九條、六十三條、六十八條和七十二條分別對突破性治療藥物、附條件批準、優先審評審批和特別審批作了相關說明(見表1)。然而,上述文件僅《疫苗管理法》提及疫苗緊急使用,而治療性藥品的緊急使用尚缺乏明確的法律依據。

表1 當前法律法規文件中涉及藥品應急或特殊審評通道的條款Tab.1 Provisions in current laws and regulations concerning drug emergency or special approval channels
2 我國現有審批制度應對突發公共衛生事件的不足
2.1 特別審批制度
2005年11月18日,原國家食品藥品監督管理局審議通過了《國家食品藥品監督管理局藥品特別審批程序》[22]。所謂藥品特別審批程序,是指在發生突發公共衛生事件時,對突發公共衛生事件應急處理所需藥品進行特別審批。該程序是突發公共衛生狀態下我國藥品應急審批的主要依據,非突發公共衛生事件不能走該審批通道。即使是應對突發公共衛生事件的藥品,也要經過嚴格篩選才能進入特別審批程序。如此次新冠肺炎疫情發生后,2020年1月22日至29日,國家藥品監督管理局藥品審評中心從企業提交的15件申請中篩選出7件進行專家組評估審核,僅通過審核3件,并報送國家藥品監督管理局啟動特別審批程序。
我國藥品特別審批制度(審批程序見圖2)不能成為突發公共衛生應急狀態下使用未上市藥品或超藥品說明書用藥的政策依據。該制度實際上是一種產品上市許可,僅加速了審批流程,但上市程序不減少,上市標準不降低,產品上市后需遵循上市后全生命周期管理的原則和規定,整個審批過程需耗費一定時間。但在突發公共衛生應急狀態下,并無足夠的時間等待產品通過特別審批后上市,因此新冠肺炎疫情期間,我國出現了使用未上市藥品或超藥品說明書用藥的情況。
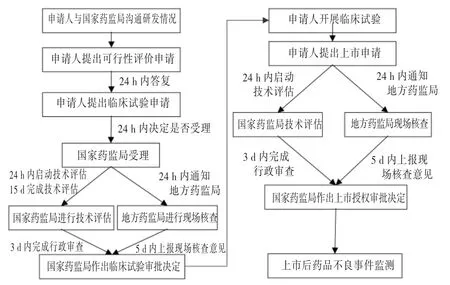
圖2 我國藥品特別審批程序Fig.2 Flow chart of special approval process of drugs in China
2.2 其他特殊審評通道或特殊用藥制度
拓展性臨床試驗制度:是指將處于臨床試驗中、用于治療嚴重危及生命且尚無其他治療手段的疾病的藥品用于未參與臨床試驗但病情相同的患者。該制度將使用人群限制在開展臨床試驗的醫療機構,且需由醫師耗費一定時間申請才能獲得,無法滿足大批人群的需求[15]。
突破性治療藥物認定:是指在臨床試驗Ⅰ期或Ⅱ期階段將具有明顯臨床優勢的創新藥或改良型新藥納入突破性治療藥物程序。該程序目的是加強溝通交流、加快臨床試驗進度,但在應急狀態下,并不能達到盡快使用的目的[21]。
附條件批準程序:允許公共衛生方面急需的藥品和應對重大突發公共衛生事件急需的疫苗在完成大部分研究程序后提前上市,屬上市途徑優化,但并不能達到足夠快速使用的目的[21]。
優先審評審批程序:適用于防治重大傳染病的創新藥和改良型新藥,也適用于疾病預防、控制所急需的疫苗。優先審評審批需在提出藥品上市許可申請時提出,其目的是加快藥品審評過程,鼓勵創新藥研發。即使進入優先審評通道,產品仍需歷經上市所需的所有程序,并不能達到足夠快速使用的目的[21]。
3 美國藥品緊急使用授權制度經驗總結
美國啟動緊急使用授權(EUA)程序應對突發公共衛生事件[23-25]。EUA是指由美國食品和藥物管理局(FDA)在緊急狀態下對未獲批準的醫藥產品及已獲批準醫藥產品的未獲批準用途進行授權。該制度經過10多年的發展,目前已較成熟,有明確的上位法、行業指南,相關文件中明確了啟動條件、授權流程、產品范圍、授權原則、授權期限和終止條件。
上位法:主要依據是《聯邦食品、藥品和化妝品法案》(簡稱《FD&C法案》)第564條,該條內容歷經多次修訂。2004年生效的《生物恐怖防疫計劃法案》對《FD&C法案》第564條進行了修訂,首次將醫藥產品緊急使用授權納入法律范疇。2013年生效的《大流行與全風險防范再授權法案》(簡稱《PAHPRA法案》)對《FD&C法案》第564條作了進一步闡述,明確了緊急使用授權的產品范圍包括藥品、生物制品和醫療器械。2016年實施的《21世紀治愈法案》再次對《FD&C法案》第564條進行了修訂,將產品范圍進一步擴大,納入動物用醫藥產品[24-25]。
行業指南:2007年7月,FDA公布了《醫藥產品緊急使用授權》文件,首次正式確定了EUA的制度程序。2017年1月,FDA發布《行業與其它利益攸關方行業指南:醫藥產品緊急使用授權與相關權限》,替代2017年發布的《醫藥產品緊急使用授權》文件[24-25]。
啟動條件和授權流程:FDA啟動緊急使用授權令的前提是國防部部長、國土安全部部長或衛生與公眾服務部部長判斷國內處于緊急狀態。整個EUA程序涉及5個步驟,1)確定是否進入緊急狀態(國防部、國土安全部或衛生與公眾服務部);2)宣布進入緊急狀態(衛生與公眾服務部);3)審評EUA的請求(FDA);4)發布EUA或拒絕EUA的請求(衛生與公眾服務部,但常會委托FDA代為發布);5)終止EUA(衛生與公眾服務部)[24-25]。詳見圖3。

圖3 美國藥品緊急使用授權流程Fig.3 Authorization process of emergency use of drugs in the United States
產品范圍及授權原則:潛在的EUA產品包括未獲批準的醫藥產品及已獲批醫藥產品未經批準的用途,此處的醫藥產品包括藥品、生物制品和醫療器械。EUA不要求醫藥產品處于特定的研發階段,但產品應正處于研發中,并完成了部分研發路徑。EUA授權必須符合以下4個法定標準:1)事態緊急;2)產品有效;3)已知潛在獲益大于潛在風險;4)無足夠的已獲批的替代產品。EUA可豁免部分監管要求,如知情同意或倫理委員會審批要求[23-25]。
授權期限和終止條件:EUA的授權期限為1年,當出現下列任一情況時,EUA隨即終止。1)緊急狀態終止時,EUA自動終止;2)EUA達1年期限時,衛生與公眾服務部部長或受其委托的FDA局長決定是否延展或終止;3)經EUA授權的產品被FDA正式許可后,對產品的EUA即可作廢[23-25]。
在H1N1流感和新冠肺炎疫情期間的應用經驗:H1N1流感期間,FDA首次將EUA授予一個未上市的研究用藥帕拉米韋,還將EUA授予磷酸奧司他韋和扎那米韋以擴大適應證。此后,美國EUA制度逐漸成熟,新冠肺炎疫情期間,FDA對多個藥品授予了EUA,截至2020年底,瑞德西韋、新冠病毒中和抗體、2種組合療法及2款新冠疫苗均已獲得EUA,這些產品在應對新冠肺炎的流行中發揮了重要作用[23]。詳見表2。

表2 美國應對H1N1流感和新冠肺炎期間授予的藥品EUATab.2 Emergency-Use-Administration(EUA)of drugs granted by the United States in response to H1N1 influenza and COVID-19
4 建立我國藥品緊急使用授權制度的建議
基于我國新冠肺炎疫情下藥品緊急使用存在的問題,借鑒美國經驗,提出如下建議。首先,完善藥品緊急使用的上位法,目前緊急使用授權的產品范圍僅包括疫苗,法律依據來自《疫苗管理法》,建議擴大緊急使用授權藥物類型,在《藥品管理法》中納入治療性藥品緊急使用授權條款。其次,在《傳染病防治法》《突發事件應對法》《藥品管理法》《疫苗管理法》中明確緊急狀態下如何啟動緊急使用授權程序。再次,基于我國現有的藥品特別審批制度,設計出藥品緊急使用授權制度程序,建議該程序可適當簡化,放寬標準,加速授權進程。最后,發布類似于行業指南的緊急使用授權指導性文件,在文件中明確緊急使用授權的啟動條件、產品范圍、授權原則、評估程序、授權期限、終止條件、責任豁免機制等內容。
猜你喜歡
中國合理用藥探索(2022年1期)2022-11-26 00:22:32
人大建設(2019年12期)2019-05-21 02:55:44
首都公共衛生(2019年5期)2019-02-12 17:32:32
瞭望東方周刊(2017年42期)2017-12-05 18:49:38
首都公共衛生(2017年1期)2017-11-29 01:21:36
環球時報(2017-03-30)2017-03-30 06:44:45
中國衛生(2016年5期)2016-11-12 13:25:28
中國衛生(2015年3期)2015-11-19 02:53:32
中國衛生(2015年5期)2015-11-08 12:09:48
中國衛生(2014年3期)2014-11-12 13:18:10
