UPLC-MS/MS同時測定特殊醫(yī)學用途配方食品中8種真菌毒素
2021-09-16 08:09:16李莉李碩
中國乳品工業(yè) 2021年8期
關(guān)鍵詞:方法
李莉,李碩
(中國食品藥品檢定研究院,北京100050)
0 引言
特殊醫(yī)學用途配方食品由于食用人群的特殊性[1],質(zhì)量安全問題一直受到消費者的高度關(guān)注。黃曲霉毒素、赭曲霉毒素和雜色曲霉素等真菌毒素,對人體具有潛在的致癌性[2],如果產(chǎn)品原料質(zhì)量和生產(chǎn)環(huán)節(jié)把控不嚴格,就有可能造成終產(chǎn)品被真菌毒素污染。目前食品中真菌毒素的檢測方法主要有酶聯(lián)免疫吸附法[3-6]、高效液相色譜法[7-10]、液質(zhì)譜聯(lián)用法[11-15]。液質(zhì)聯(lián)用和快速樣品前處理技術(shù)的聯(lián)合應用[16-19],具有高靈敏度和優(yōu)異的選擇性,應用優(yōu)勢日益突出。本文建立了直接通過式固相萃取結(jié)合UPLC-MS/MS技術(shù)檢測特殊醫(yī)學用途配方食品中8種真菌毒素的方法,該方法簡便快速,可操作性強,可以實現(xiàn)特殊醫(yī)學用途配方食品中多種黃曲霉毒素的同時快速篩查。
1 材料與方法
1.1 儀器與試劑
LCMS-8060型超高效液相色譜-三重四級桿串聯(lián)質(zhì)譜儀,日本島津公司;AL 204型分析天平,瑞士梅特勒-托利多公司;超聲波清洗器,江蘇昆山市超聲儀器有限公司;渦旋混勻器,美國Scientific Industries公司;CF 16RXII離心機,日本日立公司;MG-2200氮吹儀,日本東京理化公司;超純水純化系統(tǒng),德國賽多利斯公司。黃曲霉毒素B1(aflatoxin B1,AFB1)、黃曲霉毒素B2(aflatoxin B2,AFB2)、黃曲霉毒素G1(aflatoxin G1,AFG1)、黃曲霉毒素G2(aflatoxin G2,AFG2)、黃曲霉毒素M1(aflatoxin M1,AFM 1)、黃曲霉毒素M2(aflatoxin M2,AFM 2)混合標準溶液(10μg/mL),青島普瑞邦公司;雜色曲霉素(Sterigmatocystin,ST)標準溶液(50μg/mL),青島普瑞邦公司;赭曲霉毒素A(Ochratoxin A,OTA)標準溶液(10μg/mL),青島普瑞邦公司;乙腈(色譜純),德國默克公司;甲酸(質(zhì)譜級),F(xiàn)isher Scientific公司;Captiva EMR-Lipid萃取管(3 mL),Agilent公司;ISOLUTE Myco固相萃取柱(3 mL),Biotage公司;MPFC-QuEChERS超濾型凈化柱(高脂類),北京綠綿科技有限公司;實驗用水均為超純水;特殊醫(yī)學用途配方食品,市售。
1.2 標準溶液配制
分別準確移取AFB1、AFB2、AFG1、AFG2、AFM 1、AFM 2混合標準溶液2 mL,ST標準溶液0.2 m L,OTA標準溶液1 mL置于10 mL棕色容量瓶中,用乙腈定容至刻度,配制成AFB1、AFB2、AFG1、AFG2、AFM 1、AFM 2質(zhì)量濃度為2μg/mL、OTA和ST質(zhì)量濃度為1μg/mL的混合標準儲備溶液,-20℃避光保存。臨用時以空白基質(zhì)提取液將上述標準溶液稀釋為系列混合標準工作液。
1.3 樣品前處理
1.3.1 樣品的提取
精密稱取2.5 g(精確至0.001 g)樣品于50 mL具塞離心管中(對于加標樣品,加入所需體積的標準溶液),加入20 mL乙腈-水(80∶20,v/v)提取溶液,旋渦混勻30 s,室溫下超聲提取30 min,隨后經(jīng)8000 r/min離心5 min,取上清液待凈化。
1.3.2 樣品的凈化
移取上清液2 mL至Captiva EMR-Lipid萃取管,保持每秒1滴的流速,收集全部流出液,氮氣吹干。用0.5 mL乙腈-水溶液(50∶50,v/v)復溶,渦旋混合1 min,過0.22μm聚四氟乙烯微孔濾膜,作為待測溶液。
1.4 儀器條件
1.4.1 色譜條件
色譜柱:Aglient Infinity Poroshell 120 SB-C18(100 mm×2.1 mm,2.7μm);流動相A:A為含有0.1%甲酸的水溶液,B為含有0.1%甲酸的乙腈溶液,梯度洗脫,流速為0.3 mL/min,柱溫:40℃,洗脫程序:0~1 min,10%~20%B;1~9 min,20%~40%B;9~10 min,40%~95%B;10~14 min,95%B;14~16 min,10%B。進樣量為5μL。
1.4.2 質(zhì)譜條件
離子源:電噴霧離子源(ESI);掃描模式:正離子模式;檢測方式:多反應監(jiān)測(MRM)模式;霧化氣流量3 L/min,干燥氣流量10 L/min,加熱氣流量10 L/min,DL管溫度250℃,加熱塊溫度400℃,接口溫度300℃,接口電壓4.0 k V。其他質(zhì)譜參數(shù)如表1所示。

表1 各化合物質(zhì)譜參數(shù)
2 結(jié)果與討論
2.1 質(zhì)譜條件優(yōu)化
使用各化合物質(zhì)量濃度為100 ng/mL標準儲備液,在ESI離子源條件下采用直接進樣的方式分別進行正負離子掃描。實驗結(jié)果表明8種化合物均在正離子模式下有更好的響應,因此選擇ESI+模式。根據(jù)目標化合物的[M+H]+母離子,在多反應監(jiān)測條件下進一步優(yōu)化質(zhì)譜條件,選擇響應最強的2個子離子,得到使子離子響應最優(yōu)的三重四級桿Q 1和Q 2的電壓以及碰撞能。最終確定的質(zhì)譜多反應監(jiān)測條件參數(shù)如表1所示。
2.2 色譜條件優(yōu)化
由于8種真菌毒素是極性或者弱極性化合物,常用選用C18色譜柱,水與乙腈或甲醇混合作為流動相進行色譜分離。流動相中添加甲酸可提高目標化合物的離子化效率,提高質(zhì)譜檢測的靈敏度。本研究對比了Aglient Poroshell 120 SB-C18柱(2.1 mm×100 mm,2.7μm)、Waters BEH C18柱(2.1 mm×100 mm,1.7μm)、Waters HSS T 3(2.1 mm×100 mm,1.8μm)柱在以0.1%甲酸水溶液和0.1%甲酸乙腈為流動相時的分離效果,發(fā)現(xiàn)Aglient Poroshell 120 SB-C18柱更適合8種真菌毒素的快速分析,該柱填充2.7μm實心C18填料,其中實心內(nèi)核為1.7μm,多孔外層為0.5μm,多孔外層和實心內(nèi)核限制了擴散,提高了分離速度,而窄粒徑分布則提高了柱效和分離度,從而得到更好的分離效果和色譜峰峰型。8種真菌毒素總離子流色譜如圖1所示。

圖1 8種真菌毒素總離子流色譜
2.3 提取溶劑的選擇
根據(jù)文獻報道,黃曲霉毒素、雜色曲霉素和赭曲霉毒素常用的提取溶劑為乙腈或甲醇,在乙腈或甲醇中加入適量的水能夠促進溶劑滲透到基質(zhì)內(nèi)部,提高真菌毒素的提取效率。此外,乙腈比甲醇具有較強的蛋白沉淀能力,當樣品基質(zhì)中蛋白質(zhì)含量較多時,選取乙腈-水的混合溶液作為提取溶劑效果更好。因此,本研究以特殊醫(yī)學用途嬰兒配方食品為樣品,通過空白基質(zhì)加標的方式,考察60%乙腈水溶液、70%乙腈水溶液、80%乙腈水溶液和90%乙腈水溶液作為提取溶劑對于待測化合物回收率的影響,結(jié)果如圖2所示。由圖2可以看出,使用80%乙腈水溶液作為提取劑時8種真菌毒素的加標的回收率結(jié)果在64%~89%之間,可以滿足實驗要求。
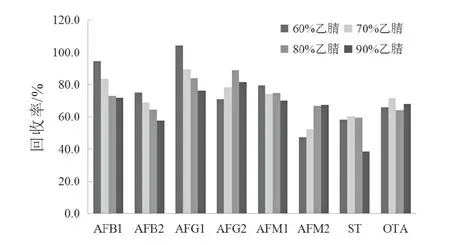
圖2 不同提取溶液對8種真菌毒素回收率的影響
2.4 樣品凈化方法的選擇
特殊醫(yī)學用途配方食品中蛋白質(zhì)、脂類和糖類等物質(zhì)含量較高,是影響質(zhì)譜測定靈敏度的主要干擾基質(zhì),因此必須通過適當?shù)膬艋椒右匀コD壳俺S玫膹碗s樣品中真菌毒素的凈化方法有免疫親和柱法、SPE、QuEChERS技術(shù)等,其中免疫親和柱價格昂貴,且只適用于單一或特定的化合物類別或樣品類型,不適合多種毒素的同時測定。本實驗對常規(guī)SPE凈化柱(ISOLUTE Myco)、直接通過式凈化柱(EMR-Lipid萃取管)和QuEChERS凈化技術(shù)(MPFC-QuEChERS)3種不同凈化方法的凈化效果進行了評價,實驗結(jié)果見圖3。分析實驗結(jié)果可以發(fā)現(xiàn),QuEChERS方法能夠高效、快速的萃取多類別的化合物,但同時也會萃取出大量基質(zhì)(尤其是脂質(zhì)),從而導致雜色曲霉素和赭曲霉毒素A基質(zhì)效應明顯、回收率偏低。常規(guī)的SPE凈化方法對于黃曲霉毒素類化合物的凈化效果要優(yōu)于直接通過式凈化柱和QuECh-ERS方法,但是操作上需經(jīng)過活化、上樣洗脫和淋洗等多個步驟,操作繁瑣,且個別目標物的加標回收率過低,不適用復雜樣品的快速分析。EMR-Lipid萃取管雖然對于黃曲霉毒素類物質(zhì)的加標回收率不如其他兩種凈化方法,但是對于雜色曲霉素和赭曲霉毒素A的加標回收率要優(yōu)于其他兩種凈化方法,綜合考慮8種目標化合物的結(jié)果,EMR-Lipid萃取管凈化方法的加標回收率均>60%,滿足實驗要求[19]。此外直接通過式凈化柱無需活化、洗脫和淋洗,可大大縮短凈化時間,因此選擇EMR-Lipid萃取管作為樣品前處理的凈化方法。

圖3 不同凈化方法對8種真菌毒素回收率的影響
2.5 方法研究及結(jié)果分析
2.5.1 線性范圍、回歸方程和檢出限
在質(zhì)譜分析中,基質(zhì)效應會嚴重影響痕量水平目標化合物定量分析的準確性[20]。為了補償基質(zhì)效應對檢測結(jié)果帶來的影響,本研究采用基質(zhì)匹配標準曲線進行目標化合物定量分析。將不含待測物的空白樣品按照1.3步驟進行處理得到空白基質(zhì)溶液,用空白基質(zhì)溶液稀釋混合標準儲備液得到系列濃度的基質(zhì)加標溶液,上機測定。以8種真菌毒素的質(zhì)量濃度(ng/mL)為橫坐標,定量離子峰面積為縱坐標,繪制標準曲線。計算目標化合物信號響應和噪聲的比值S/N,得到方法檢出限(limit of detections,LOD,S/N=3)和定量限(limit of quantifications,LOQ,S/N=10),結(jié)果如表2所示。6種黃曲霉毒素在0.5~20 ng/mL質(zhì)量濃度范圍內(nèi)、雜色曲霉素和赭曲霉素A在0.25~10 ng/mL質(zhì)量濃度范圍內(nèi)呈線性關(guān)系,線性相關(guān)系數(shù)r>0.992,8種真菌毒素的檢出限為0.06~2.1μg/kg,定量限為0.2~2.9μg/kg。
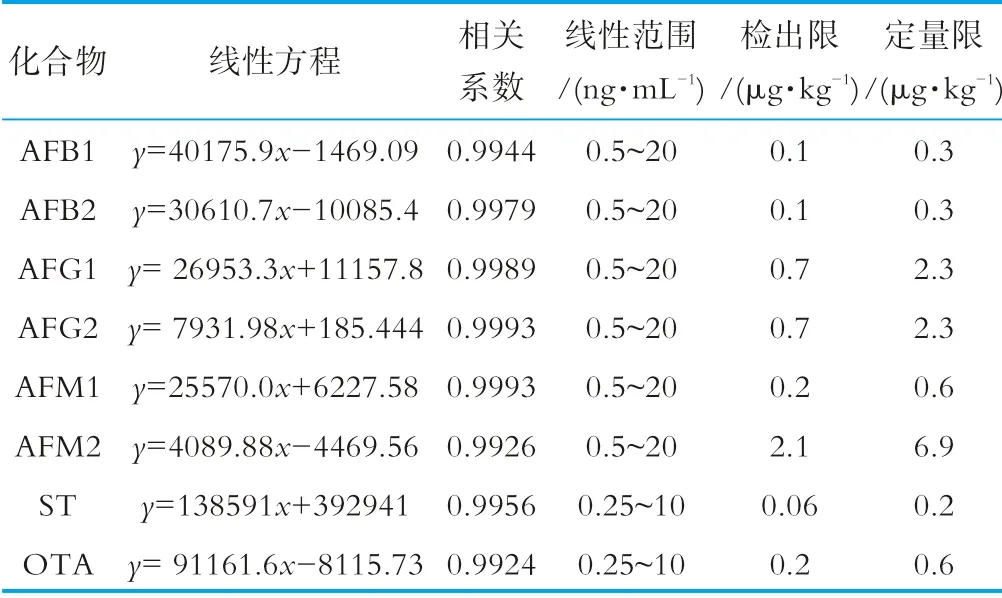
表2 8種真菌毒素的線性范圍、相關(guān)系數(shù)、檢出限和定量限
2.5.2 方法的回收率及精密度
向空白樣品添加低、中、高3個濃度水平的標準溶液進行加標回收試驗,按照方法給出的步驟進行樣品提取、凈化及上機檢測,計算目標化合物的加標回收率。每個加標濃度平行測定6份,用測定結(jié)果的相對標準偏差評價精密度(relative standard deviation,RSD)。回收率和精密度結(jié)果如表3所示。由表3可以看出,8種真菌毒素的平均回收率為92.2%~108.9%,RSD小于15%。
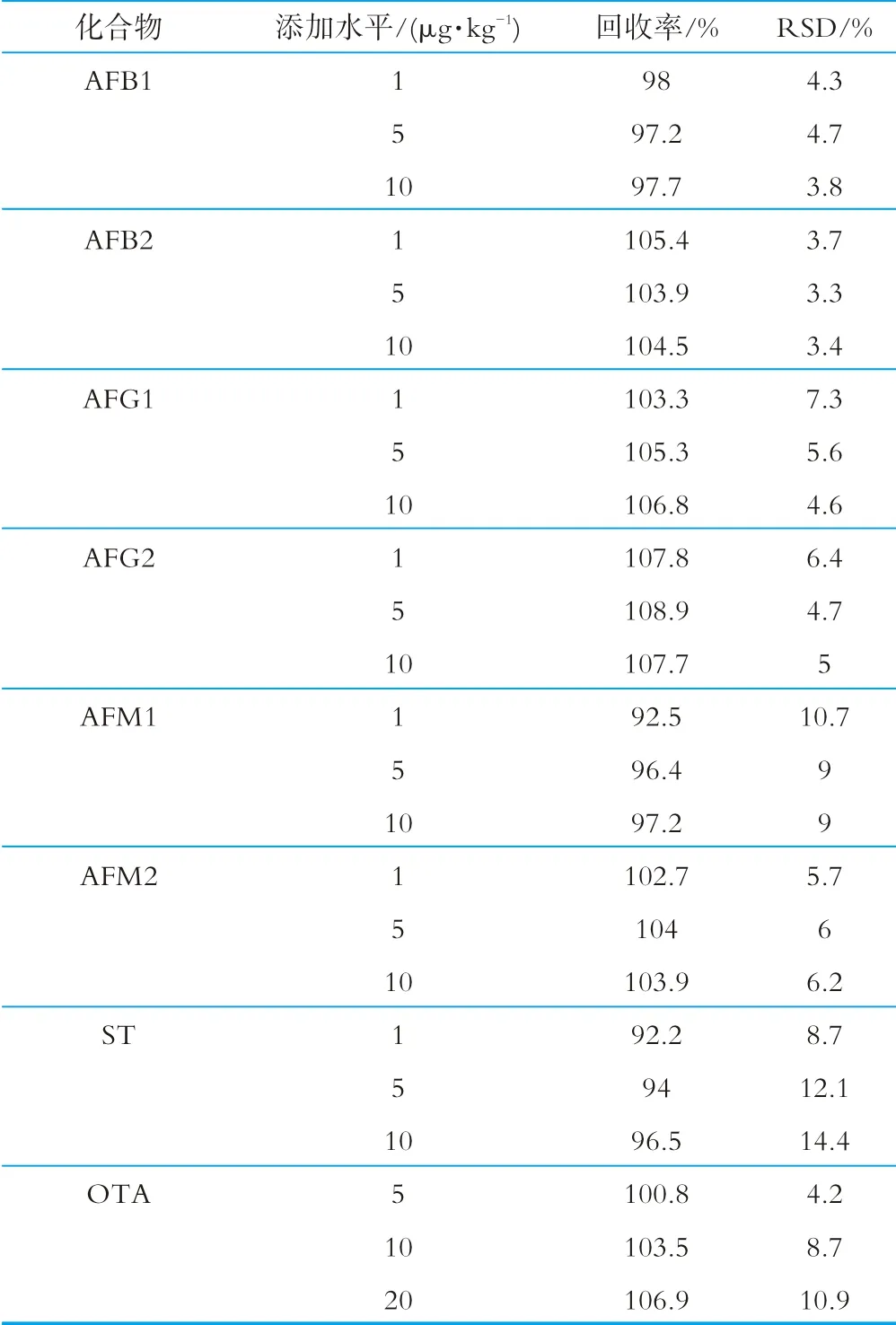
表3 回收率和精密度實驗結(jié)果(n=6)
2.5.3 實際樣品檢測
按照本實驗建立的方法對購自不同產(chǎn)地和廠家的15份特殊醫(yī)學用途配方食品進行檢測,15批樣品中均未檢出8種真菌毒素。
3 結(jié) 論
本研究建立了采用新型固相萃取前處理方法結(jié)合液相色譜質(zhì)譜聯(lián)用檢測技術(shù)同時測定特殊醫(yī)學用途配方食品中多種類真菌毒素的快速篩查方法。直接通過式EMR-Lipid萃取管省去了常規(guī)萃取柱活化、洗脫和淋洗的步驟,可大大縮短凈化時間,提高檢測的效率,可以實現(xiàn)批量樣品的快速檢測。該方法簡化了分析步驟,操作簡便快捷,同時具有良好的準確度和精密度,能滿足特殊醫(yī)學用途配方食品中真菌毒素污染水平的監(jiān)測和產(chǎn)品質(zhì)量控制工作的需要。
猜你喜歡
中老年保健(2021年9期)2021-08-24 03:52:04
河北畫報(2021年2期)2021-05-25 02:07:46
中學生數(shù)理化(高中版.高考理化)(2020年2期)2020-04-21 05:33:04
兒童繪本(2020年5期)2020-04-07 17:46:30
兒童故事畫報(2019年5期)2019-05-26 14:26:14
Coco薇(2016年2期)2016-03-22 02:42:52
山東青年(2016年1期)2016-02-28 14:25:23
Coco薇(2015年1期)2015-08-13 02:47:34
小雪花·成長指南(2015年7期)2015-08-11 15:03:12
小雪花·成長指南(2015年4期)2015-05-19 14:47:56
