超高效液相色譜法測定濃縮飼料中維生素K3含量的不確定度評定
2021-10-23 12:44:26畢融冰王玉方孫紫薇金美伶劉繼永
分析儀器 2021年3期
畢融冰 趙 卉 王玉方 孫紫薇 金美伶 劉繼永
(中國農業科學院特產研究所, 長春 130000)
測量不確定度(measurement uncertainty,MU),簡稱不確定度,是根據所用到的信息,表征賦予被測量值分散性得的非負參數[1]。不確定度一般由若干分量組成,其中一些分量可以根據一系列測量值的統計分布進行評定(A類測量不確定度);另一些分量則可以根據經驗或其它信息獲得的概率密度函數進行評定(B類測量不確定度)[2]。通過比較測定過程中各分量引入的相對標準不確定度,并對產生不確定度的主要來源進行分析,可以找出對檢測結果產生較大影響的因素并加以控制[3-5]。
維生素K為肝臟合成酶的必須物質,并參與骨骼素的合成,防止畜禽體質軟弱、皮下出血,對動物生長發育起著重要的作用,適當補充維生素K,可以避免維生素K缺乏癥,因此被廣泛用作飼料添加劑。《飼料添加劑品種目錄(2013)》[6]收錄了亞硫酸氫鈉甲萘醌(MSB)、亞硫酸氫煙酰胺甲萘醌(MNB)、二甲基嘧啶醇亞硫酸甲萘醌(MPB)三種維生素K3類飼料添加劑,三者均以甲萘醌為官能團[7]。目前飼料中維生素A、D3、E和B2的不確定度研究已見報道[5,8-10],張艷紅等[5]利用HPLC法對預混合飼料中維生素A、維生素D3和維生素E的測定進行不確定度評定,結果表明:采用最小二乘法求得樣品質量濃度的過程和標準溶液配置過程中引入的不確定度貢獻較大;陳楓等[10]利用HPLC法對預混合飼料中維生素B2的測定進行不確定度評定,結果表明:重復性實驗和標準溶液曲線擬合中引入的不確定度貢獻較大;而飼料中維生素K3的不確定度研究較少,本文依據國家標準GB/T18872-2017和JJF1059.1-2012國家計量技術規范,建立數學模型,對UPLC法測定濃縮飼料中維生素K3的含量進行不確定度的評定,以期找出對測量結果影響較大的因素,為濃縮飼料中維生素K3的質量控制提供理論依據。
1 材料與方法
1.1 材料與試劑
三氯甲烷、無水碳酸鈉、無水硫酸鈉、硅藻土(均為分析純);甲醇(色譜純,美國Fisher公司);甲萘醌標準品(98.08 %,德國Dr.Ehrenstorfer);試驗用水均為一級水;濃縮飼料(隨機選取市售濃縮飼料)。
1.2 儀器與設備
超高效液相色譜儀(Acquity UPLC H-Class,美國Waters公司),配有二極管陣列(PDA)檢測器;Mettler-Toledo MS204S 電子天平(d=0.0001 g),Mettler-Toledo XS205電子天平(d=0.00001 g);高速離心機(安徽中科中佳科學儀器有限公司);氮吹儀(上海安譜實驗科技股份有限公司);調速多用振蕩器(江蘇省金壇市榮華儀器制造有限公司)。
1.3 試驗方法
1.3.1標準溶液的制備
稱取甲萘醌標準品25 mg(精確至0.00001 g)于50 mL棕色容量瓶中,甲醇溶解并定容至刻度,混勻,為甲萘醌標準儲備液,-18 ℃保存。準確吸取0.50 mL甲萘醌標準儲備液于50 mL棕色容量瓶中,甲醇溶解并定容至刻度,混勻,為甲萘醌標準工作液,工作液濃度約為5 μg·mL-1,-18 ℃保存。
1.3.2試樣溶液的制備
濃縮飼料過60目篩,混勻,稱取5 g(精確至0.0001 g)于100 mL具塞錐形瓶中,準確加入50 mL三氯甲烷,放在調速多用振蕩器上振蕩2 min;加入5 mL 1 mol·L-1碳酸鈉溶液,振蕩3 min;加入5 g硅藻土和無水硫酸鈉混合物(3:20,g/g),振蕩30 min;5000 r·min-1,離心10 min。
依據預實驗確定分取量,準確吸取20 mL三氯甲烷提取液,氮氣吹干,甲醇溶解,定容至5 mL棕色容量瓶中,通過0.22 μm有機系濾膜,用于UPLC分析。
1.3.3色譜條件
色譜柱:ACQUITY UPLC BEH C18反相色譜柱(2.1×100 mm,1.7 μm美國Waters公司);流動相:甲醇-水(75∶25,V/V);柱溫:室溫;流速:0.4 mL·min-1,檢測波長:251 nm;進樣量:3 μL。
1.3.4定量測定
依次注入相應的甲萘醌標準工作液和試樣溶液,根據色譜峰面積的響應值,用外標法定量測定。
2 測量不確定度的評定
2.1 數學模型
維生素K3的含量計算:
(1)
式(1)中:X為濃縮飼料中甲萘醌的含量,mg·kg-1;A1為濃縮飼料試樣溶液峰面積值;V1為提取液總體積,mL;V3為定容體積,mL;ρ為甲萘醌標準工作液濃度,μg·mL-1;A0為甲萘醌標準工作液峰面積值;m為濃縮飼料質量,g;V2為提取液分取體積,mL。
2.2 不確定度來源分析
濃縮飼料中維生素K3含量測定的不確定度來源如圖1所示。

圖1 濃縮飼料中維生素K3含量測定的不確定度來源
由圖1,對濃縮飼料中維生素K3含量測定結果有影響的各種不確定度分量來源進行分析,主要有:標準物質產生的不確定度,包括標準物質本身、標準物質稱量、配制標準儲備液和標準工作液所產生的不確定度;試樣溶液產生的不確定度,包括試樣稱量、試樣溶液制備過程所產生的不確定度;樣品處理操作過程的差異,即回收率產生的不確定度;儀器測量重復性產生的不確定度,包括試樣溶液峰面積和標準品峰面積產生的不確定度。
2.3 不確定度分量的評定
2.3.1標準物質產生的不確定度
標準物質產生的不確定度包括標準物質、標準物質稱量、配制標準儲備液和標準工作液產生的不確定度,屬于B類不確定度。
2.3.1.1標準物質引入的不確定度
甲萘醌標準品由德國Dr.Ehrenstorfer公司生產,由標準物質證書可知其擴展不確定度為0.56 %(k=2),純度為98.08 %,則相對標準不確定度為:
2.3.1.2標準物質稱量引入的不確定度
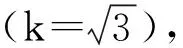
相對標準不確定度為:
2.3.1.3標準儲備液和標準工作液配制過程中引入的不確定度
稱取甲萘醌標準品于50 mL棕色容量瓶中,甲醇溶解并定容至刻度,混勻,為標準儲備液;使用0.5 mL分度吸量管準確吸取0.5 mL標準儲備液于50 mL棕色容量瓶中,甲醇溶解并定容至刻度,混勻,得到濃度為5 μg·mL-1甲萘醌標準工作液。


表1 標準儲備液和標準工作液配制過程產生的不確定度
則由標準儲備液和標準工作液配制過程產生的相對標準不確定度為:
=0.0186
由上述不確定度分量可以得到甲萘醌標準物質產生的相對標準不確定度:
2.3.2試樣溶液產生的不確定度
試樣溶液產生的不確定度包括試樣稱量、試樣溶液制備過程產生的不確定度,屬于B類不確定度。
2.3.2.1試樣稱量產生的不確定度
相對標準不確定度為:
2.3.2.2試樣溶液制備過程產生的不確定度
稱取5 g試樣于100 mL具塞錐形瓶中,用50 mL單標線吸量管準確加入50 mL三氯甲烷,振蕩2 min;用5 mL分度吸量管加入5 mL碳酸鈉溶液,振蕩3 min;5000 r/min,離心10 min。用20 mL單標線吸量管準確吸取20 mL提取液,氮氣吹干,甲醇溶解,定容至5 mL棕色容量瓶中,備用。

表2 試樣溶液制備過程產生的不確定度
則試樣溶液制備過程產生的相對標準不確定度為:
=0.00773
由上述不確定度分量可以得到試樣溶液制備過程產生的相對標準不確定度:
2.3.3儀器重復測量產生的不確定度
2.3.3.1標準品峰面積(A0)重復測量產生的不確定度
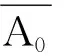
則標準品峰面積(A0)重復測量產生的相對標準不確定度為:
2.3.3.2試樣溶液峰面積(A1)重復測量產生的不確定度

則試樣溶液峰面積(A1)重復測量產生的相對標準不確定度為:
由計算公式計算試樣溶液中維生素K3的含量:
2.3.4試樣中添加回收率產生的不確定度
則試樣中添加回收率產生的相對標準不確定度為:
用t檢驗法來考察回收率R與1之間是否具有顯著性差異:
t值大于雙側臨界值t(95,5)=2.571,則回收率R與1之間具有顯著性差異,需要回收率R校正測定結果,則試樣溶液中維生素K3的含量為:
2.4 合成相對不確定度和擴展不確定度的計算
各分量的相對標準不確定度見表3。

表3 各分量的相對標準不確定度
則合成相對不確定度為:

取包含因子k=2(置信水平為95%),則測得試樣溶液中維生素K3的相對擴展不確定度為:
U(X)=k·urel(X)=2×0.0233=0.0466
2.5 結果報告
經回收率R校正,試樣溶液中維生素K3的含量為1.42 mg·kg-1,則其擴展不確定度為:
U=U(X)·X=0.0466×1.42=0.07mg·kg-1
故本試驗濃縮飼料中維生素K3的UPLC法測定結果為:1.42±0.07mg·kg-1,k=2(95%置信度)。
3 分析
由圖2可知,UPLC測定濃縮飼料中維生素K3含量的過程中對不確定度影響最小的是urel(A0),其次是urel(R)、urel(s)和urel(A1),urel(r)是影響本次試驗結果的主要來源,由圖3可知,標準物質產生的不確定度分量中,urel(Vr)是主要影響因素。

圖3 標準物質各分量產生的相對標準不確定度
綜上,配制標準儲備液和工作液過程中,適當增大分取體積,使用經過檢定/校準合格的量器,避免實驗操作過程中實驗室溫度波動過大,可以減小由標準物質引入的不確定度。實驗涉及的重要儀器設備應定期檢定/校準和維護,保證儀器設備狀態,降低由儀器設備引入的不確定度。通過避光操作,減小實驗過程中甲萘醌遇光分解產生的損失,減小添加回收率產生的不確定度。通過以上實驗操作,來減小各分量產生的不確定度,進而達到提高檢測結果準確性和可靠性的目的,為濃縮飼料中維生素K3的質量控制提供理論依據。
猜你喜歡
城市道橋與防洪(2022年4期)2022-07-01 06:04:12
中學生數理化·八年級物理人教版(2019年9期)2019-11-25 07:33:02
當代陜西(2019年8期)2019-05-09 02:22:48
中學生數理化·八年級物理人教版(2019年3期)2019-04-25 06:20:54
動漫星空(興趣百科)(2019年3期)2019-03-07 07:23:10
中學生數理化·八年級物理人教版(2018年3期)2018-05-31 08:52:45
家庭影院技術(2018年4期)2018-05-09 07:07:52
數學小靈通(1-2年級)(2017年10期)2017-11-08 08:39:45
少兒科學周刊·兒童版(2016年1期)2016-03-14 03:52:21
專用汽車(2016年4期)2016-03-01 04:13:43
