基于生信分析ATRA治療APL后的關鍵差異基因
2021-11-02 07:00:26羅斯琦婁世鋒
世界最新醫學信息文摘 2021年67期
羅斯琦,婁世鋒
(重慶醫科大學附屬第二醫院血液內科,重慶 400010)
1 研究背景與思路分析
急性早幼粒細白血病(acute promyelocytic leukemia,APL)是一種以骨髓分化在早幼粒細胞階段停滯導致細胞蓄積為特征的急性髓系細胞白血病(acute myelocytic leukemia,AML),在WHO分型中屬M3型急性髓系白血病(AML-M3),APL患者t(15;17)(q22;q21)染色體易位產生PML-RARα融合基因[1],該融合基因是導致APL中骨髓細胞分化停滯的主要機制[2]。全反式維甲酸(all-trans-retinoic acid,ATRA)聯合三氧化二砷對APL患者的誘導分化治療是目前常用的靶向治療藥物,治愈率高,可以將完全緩解(completeremission,CR)率 提 高 到90%[3],ATRA在APL細 胞 中 誘 導 分 化 的 機制包括:(1)ATRA將PML-RARa從轉錄阻遏物轉化為轉錄激活物[4];(2)ATRA通過半胱氨酸蛋白酶或泛素/蛋白酶體系統降解PML-RARa融合蛋白,同時釋放野生型RXR和PML,并誘導PMLNBs重組,PML/RARalpha直接參與賦予APL細胞ATRA敏感性,并 且ATRA誘導的PMLNBs重 組是PML/RARalpha消 失 的 結 果[5];(3)RAF-1/MEK/ERK信 號通路被激活并通過調節C/EBPb,C/EBPe和PU.1的蛋白質水平來調節ATRA誘導的APL細胞分化[6],第四為一個非基因組途徑,ATRA激活PKA從而導致ATRA靶基因的轉錄增加和APL的分化[7]。在ATRA治療過程中,可能出現分化綜合征(Differentiationsyndrome,DS),又稱維甲酸綜合征(Retinoicacidsyndrome,RAS)[8],同時仍有ATRA治療后復發的情況發生,減少APL患者的早期死亡率以及減少復發率,是APL治療的研究熱點。因此,本研究旨在通過生物信息學工具分析ATRA處理APLNB4細胞(一種從人急性早幼粒細胞白血病(M3)中分離的具有t(15;17)標記的成熟誘導細胞系,NB4)前后基因表達譜差異,并尋找差異基因富集的信號途徑,探討全反式維甲酸治療急性早幼粒細白血病的可能分子作用機制,同時篩選出信號通路中高表達提示預后不良的關鍵基因,為尋找APL治療的新靶點提供思路。
2 資料和方法
2.1 一般資料
以美國國立生物技術信息中心(NCBI)的高通量基因表達數據庫(GEO)為工具,以“all-trans-retinoic acid”為關鍵詞,選取并下載下載ATRA分化的野生型和TG2基因敲除NB4細胞的基因表達譜數據集(GSE23702),其來源于德布勒森大學,采用平臺為GPL6244[HuGene-1_0-st] Affymetrix Human Gene 1.0 ST Array [transcript (gene) version]。選取其中ATRA處理前、48h、72h后的野生型APL NB4細胞基因表達數據,每個時間點選取3個樣本。
2.2 方法
2.2.1 基因芯片質量驗證
使用GEO2R(https://www.ncbi.nlm.nih.gov/geo/geo2r) R boxplot對表達式數據應用分位數生成箱線圖,如果若某個芯片的箱線圖出現較大偏差則表明該芯片存在問題。使用R limma (plotDensities)生成表達密度圖,如果圖中曲線相差偏離大,則在差異表達分析之前進行數據歸一化。使用limma(qqt) 生成t統計分位線圖評估limma測試結果的質量。
2.2.2 差異基因篩選
在GEO2R使 用Bioconductor項 目 中 的GEOquery和limma R包對分析篩選GEO數據集中不同數據組的差異表達水平[9]。在GSE23702數據集中以未經過ATRA處理過的野生型APL NB4細胞基因表達數據(GSM594067、GSM594068、GSM594069)為對照組分別與ATRA處理48h(GSM594073、GSM594074、GSM594075)、72h(GSM594079、GSM594080、GSM594081)后的野生型APL NB4細胞基因表達數據對比,篩選標準為矯正后P<0.05并且l log(Fc)I>2,使用GEO2R在線工具limma(volcanoplot)包生成火山圖,并顯示統計顯著性(-log10P值)與變化幅度(log2倍變化)的對比,使差異基因可視化,最后使用limma(vennDiagram)包生成維恩圖,用于探索和下載多個對比之間重要基因的重疊,可聯合使用在線工具Draw Venn Diagram(http://bioinformatics.psb.ugent.be/webtools/Venn/)篩選出在ATRA處理48h和72h后同時上調或下調的DEGs用于進一步分析。
2.2.3 基因本體論(gene ontology,GO)富集分析和京都基因與基因組百科全書(Kyotoencyclopedia of genes and genomes,KEGG)通路篩選
用DAVID 6.8在線數據庫(https://david.ncifcrf.gov/tools.jsp)對DEGs進行基因本體論(gene ontology,GO)注釋和京都基因與基因組百科全書(Kyotoencyclopedia of genes and genomes,KEGG)通路分析[10],聯合基于KEGG的注釋系統KOBAS(http://kobas.cbi.pku.edu.cn/kobas3/?t=1)尋找差異基因富集的關鍵信號通路[11]。
2.2.4 PPI網絡構建和分析
使 用String在 線 數 據 庫(https://string-db.org/)對 上 述DEGs進行分析,構建PPI網絡[12]后下載并使用Cytoscape(Version3.8.2)軟件進行可視化[13],利用cytoHubba插件分析預測該網絡中最高相關度的前10個DEGs作為ATRA誘導細胞分化相關的關鍵基因,即hub基因[14]。
2.2.5 統計學分析
運用GEO2R工具的Benjamini & Hochberg法分析GSE23702表達譜芯片數據,采用SAM法篩選芯片數據中DEGs。在DAVID數據庫中,通過Bonferroni 校正法和Bootstrap 法等進行GO分析的檢驗,利用Fisher精確概率檢驗和基因富集分析等方法進行KEGG分析。在Cytoscape中,選取CytoHubba插件中MCC拓撲分析算法進行數據分析。
3 結果
3.1 芯片質量驗證
用GEO2R對表達式數據生成箱線圖(圖1),其中以中位數為中心的值表示數據已標準化且可交叉比較;使用R limma(plotDensities)生成表達密度圖(圖2),圖中9條曲線之間趨勢相對平行,使用limma(qqt) 生成t統計分位線圖(圖3),其中圈基本沿一條直線走行,說明limma測試結果的質量較好,該芯片質量優,檢測結果穩定,可為后續分析提供可靠數據。
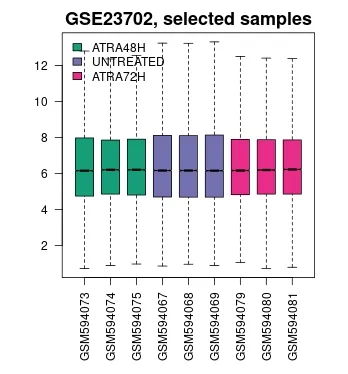
圖1 箱線圖
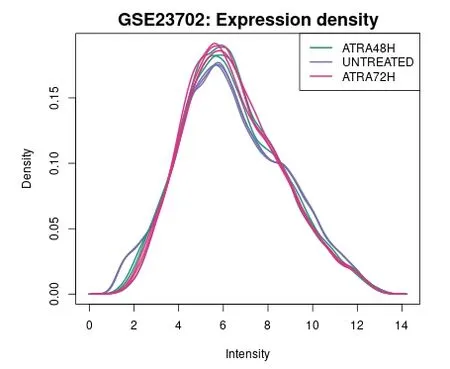
圖2 表達密度圖

圖3 t統計分位線圖
3.2 DEGs篩選
用GEO2R使用limma(vennDiagram)對3組數據:ATRA處理 前(UNTREATED)、48h(ATRA48H)、72h(ATRA72H)后 的 野 生型APL NB4細胞基因表達數據進行綜合分析(設置P<0.05)并生成韋恩圖(圖4),可以看出ATRA處理48h后(ATRA48H)與未處理組的差異基因與處理72h(ATRA72H)后與未處理組的差異基因有相同部分,但ATRA處理48h與ATRA處理72h組無差別,因此我們分別選取ATRA48H、ATRA72H與UNTREATED組進行差異分析,用P<0.05并且l log(Fc)I>2進行篩選;與未用ATRA處理相比ATRA處理48h后篩選出上調基因140個,下調基因96個,共計236個 DEGs;ATRA處理72h后篩選出上調基因191個,下調基因131個,共計322個 DEGs,結果分別用火山圖(圖5和圖6)進行展示,上調和下調最顯著的前10個DEGs具體信息分別整理為表格(表1和表2),同時兩組數據上調和下調最顯著的前10個 DEGs取交集得出韋恩圖(圖7和圖8)。
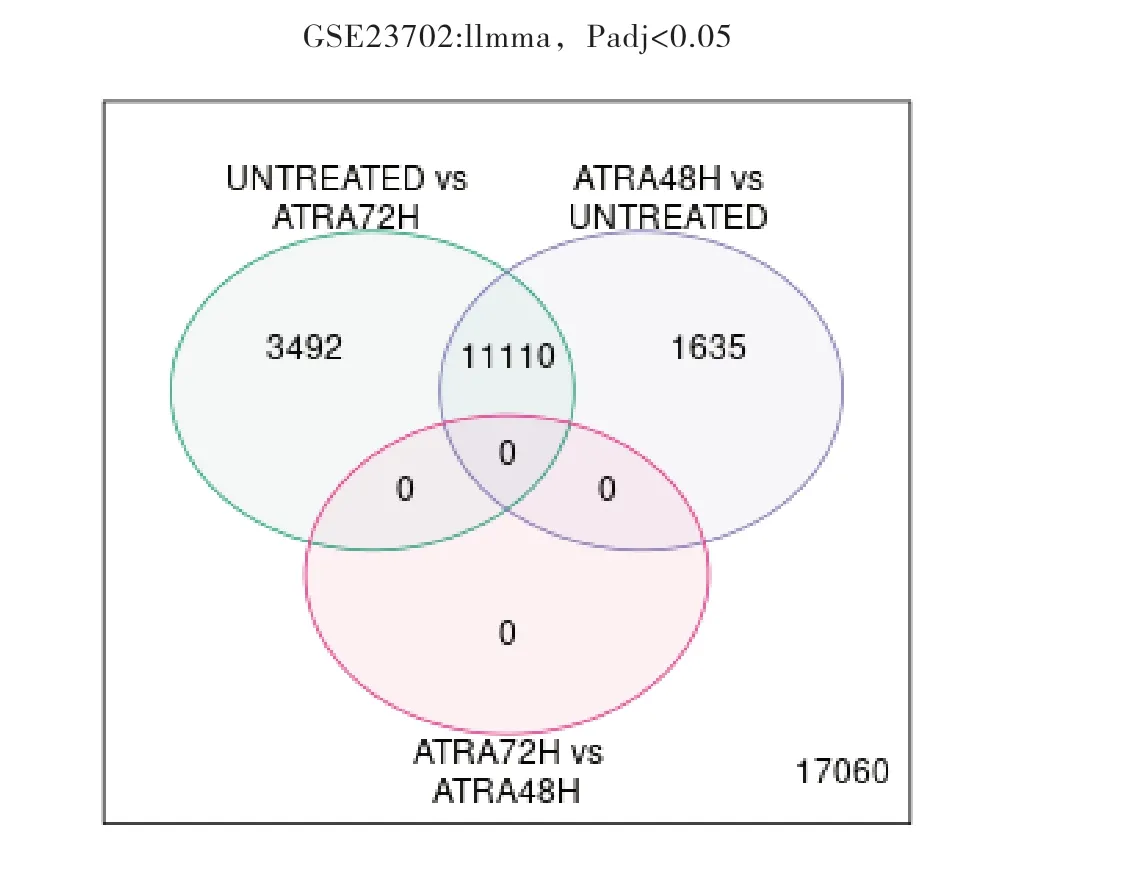
圖4 ATRA處理前(UNTREATED)、48h(ATRA48H)、72h(ATRA72H)后的野生型APL NB4細胞基因表達交集數據韋恩圖
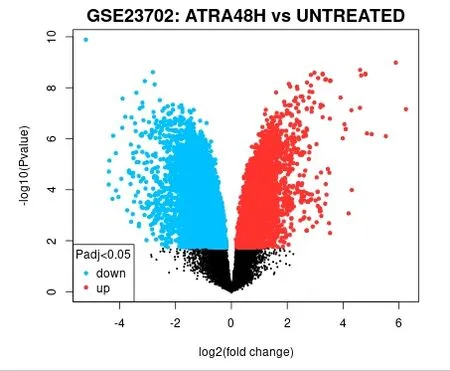
圖5 與未用ATRA處理相比ATRA處理48h后基因分析火山圖,黑色代表非 DEGs,紅色代表上調 DEGs,藍色代表下調 DEG

圖6 與未用ATRA處理相比ATRA處理72h后基因分析火山圖,黑色代表非 DEGs,紅色代表上調 DEGs,藍色代表下調 DEG
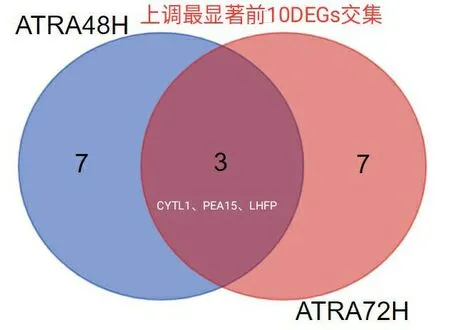
圖7 ATRA分別處理48h和72h后上調最顯著的前10個 DEGs取交集

圖8 ATRA分別處理48h和72h后下調最顯著的前10個 DEGs取交集
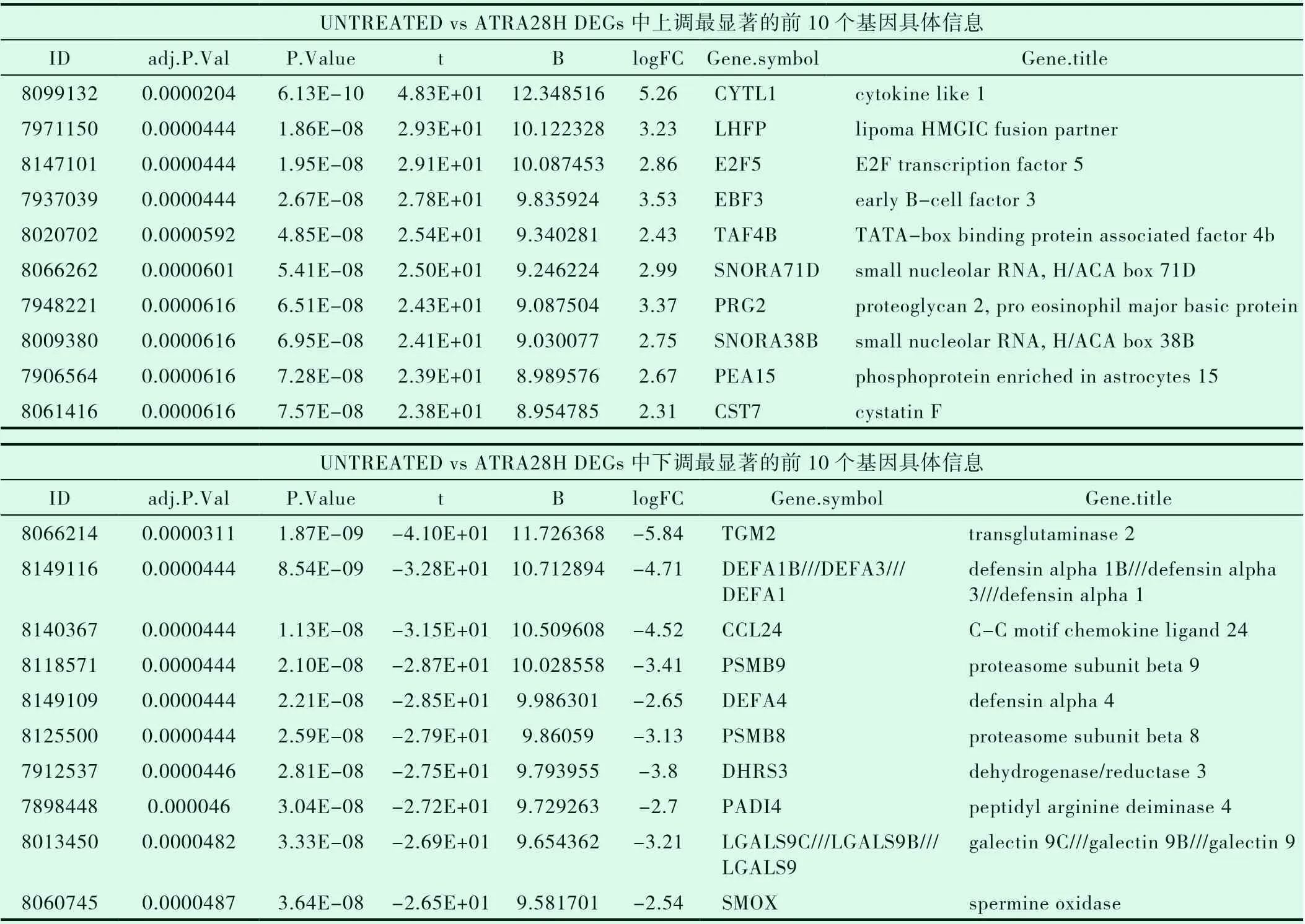
表1 UNTREATED vs ATRA48H DEGs中上、下調最顯著的前10個基因具體信息
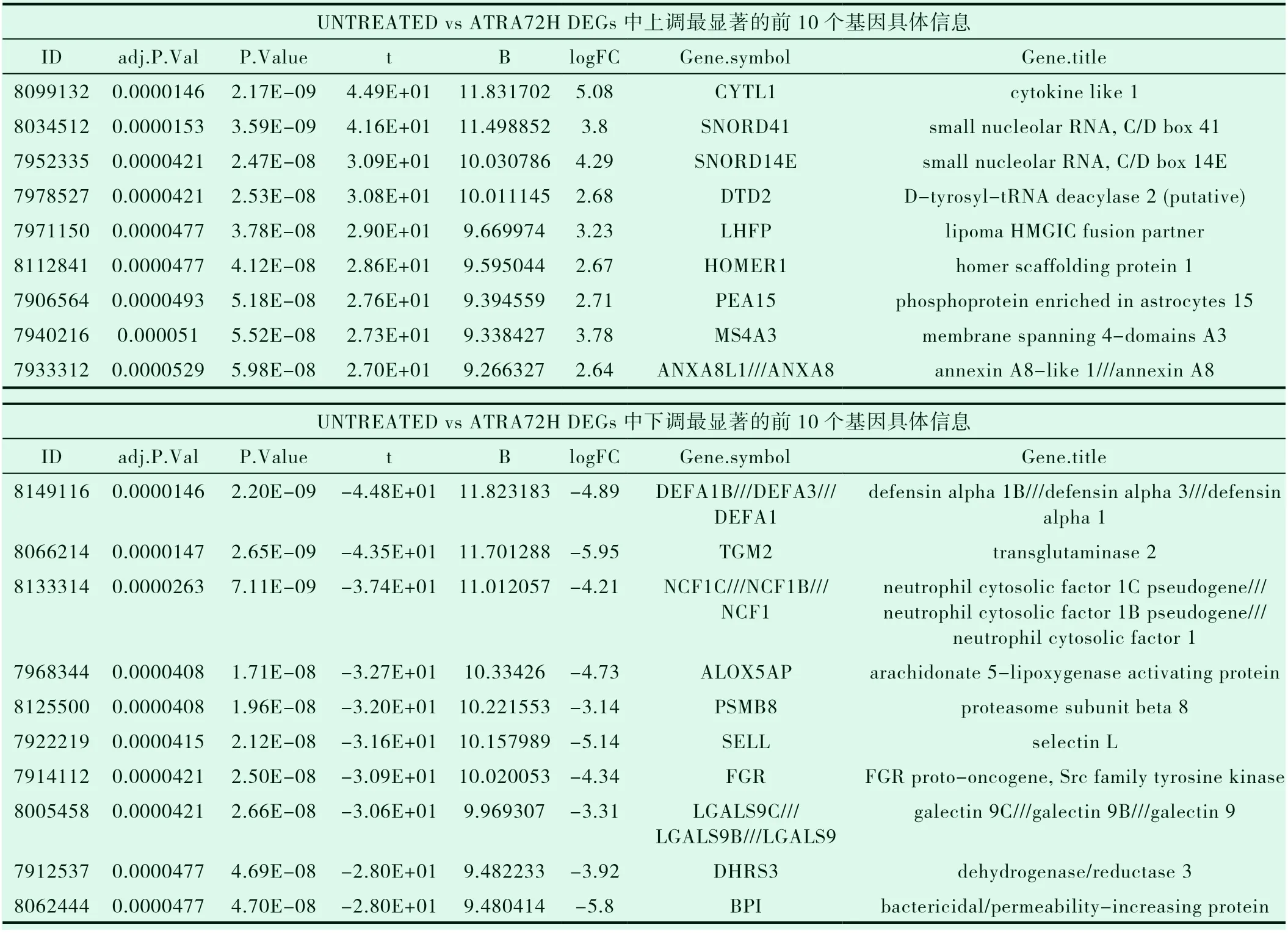
表2 UNTREATED vs ATRA72H DEGs中上、下調最顯著的前10個基因具體信息
3.3 DEGs的GO 和KEGG富 集 分 析
DEGs的GO富集分析結果用柱狀圖(圖9和10)表示如下,結果表明ATRA48H后上調DEGs在生物過程(biological process,BP)中主要與一氧化氮的生物合成過程、細胞周期調控、凋亡過程的負調控、葡萄糖進入細胞的負調控、異源代謝過程、RNA聚合酶II轉錄預起始復合物裝配、建立平面極性、宿主共生體生長的負調控、腎臟發育等過程相關;在細胞組成(cellular component,CC)層面主要與細胞纖維網、分泌顆粒、細胞的頂端部分等結構相關;在分子功能(Molecular Function,MF)上主要與苦味受體活性、轉錄因子活性,RNA聚合酶Ⅱ核心啟動子序列特異性結合參與預啟動復合物組裝、碳酸鹽脫水酶活性、味覺受體活性等分子功能相關。ATRA48H后下調DEGs在BP中主要與對抗原刺激的急性炎癥反應、激活先天免疫反應、B細胞增殖的負調控、白三烯代謝過程、凋亡細胞清除、干擾素-γ介導和整合素的信號通路、中性粒細胞趨化途徑、細胞粘附、調節細胞形狀、白細胞遷移、細胞防御反應、細胞對干擾素-β的反應途徑、激活先天免疫反應、蛋白質結合的正調控、經由TAP依賴的MHC I類進行抗原加工和呈遞外源肽抗原、細胞表面受體信號通路、炎癥反應的正調控、血管生成的正調控、Wnt信號通路的正調控、蛋白質瓜氨酸化等過程相關;在CC層面主要與細胞外泌體、胞漿、細胞膜與質膜、蛋白酶體核心復合體、精原蛋白酶體復合物等結構相關;在MF上主要與細胞粘附分子、鈣離子、整聯蛋白、花生四烯酸結合,蛋白質精氨酸脫亞氨酶、蘇氨酸型內肽酶活性等分子功能相關。用同樣的方法分析ATRA72H后上調和下調DEGs與ATRA48H差別不大,選取主要富集通路用柱狀圖(圖11和12)顯示。結果用KEGG可視化分析(篩選條件為校正后P<0.05)結果表明,ATRA48H上調DEGs主要與流體剪切應力與動脈粥樣硬化條信號及代謝通路(hsa05418)密切相關,與下調DEGs相關的通路共篩選出15條,具體信息顯示在表3中,其中包括趨化因子信號通路(hsa04062)、細胞粘附分子(hsa04514)、NOD樣受體信號轉導通路(hsa04621)、白細胞跨內皮遷移(hsa04670)、碳水化合物消化和吸收(hsa04973)、PI3K-Akt信號通路(hsa04151)、造血細胞譜系(hsa04640)、蛋白酶體(hsa03050)等。用同樣的方法分析ATRA72H后上調和下調DEGs與ATRA48H差別不大,結果顯示在表4中。
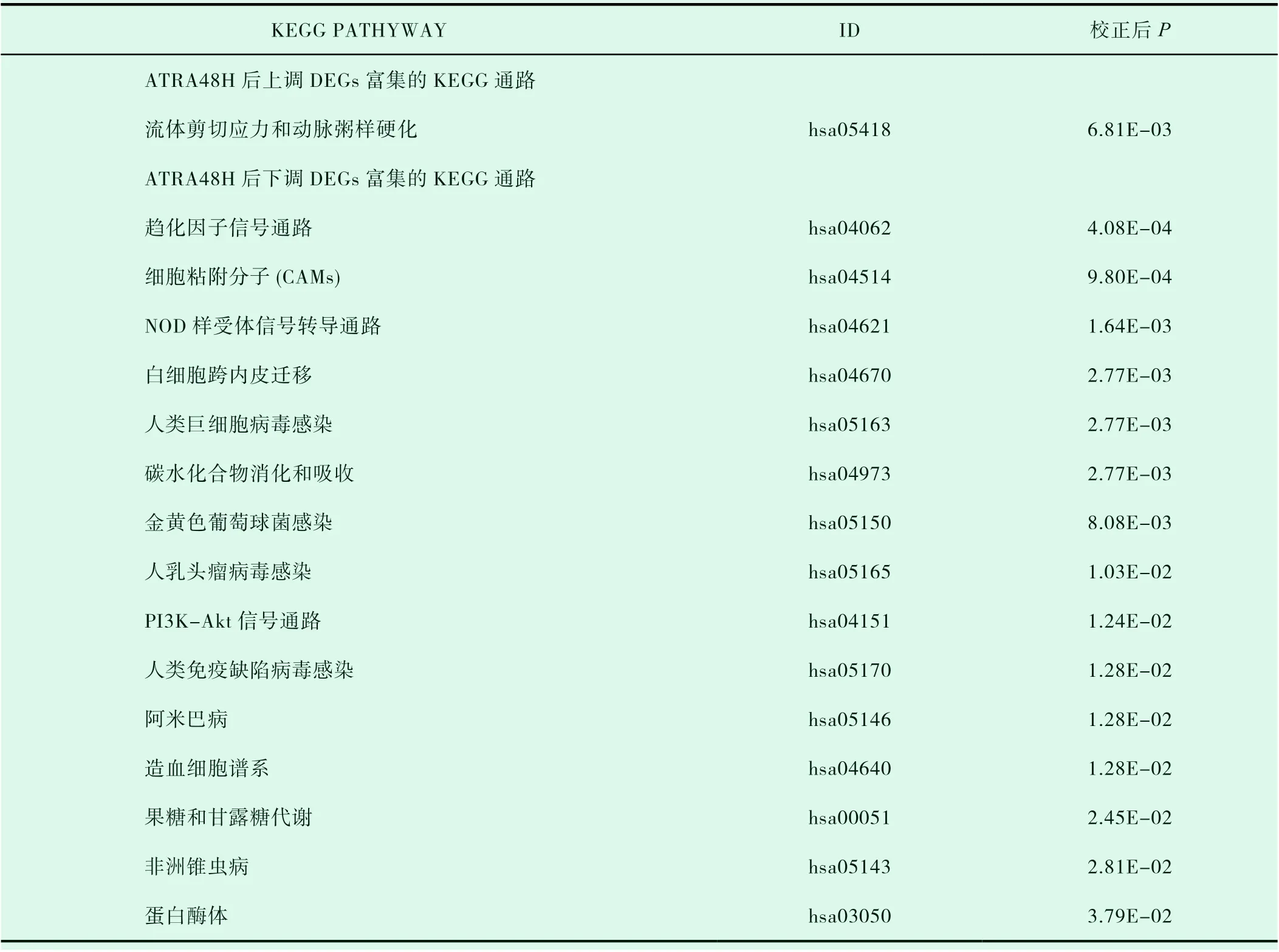
表3 UNTREATED vs ATRA48H 上、下調DEGs通過KEGG分析的主要通路
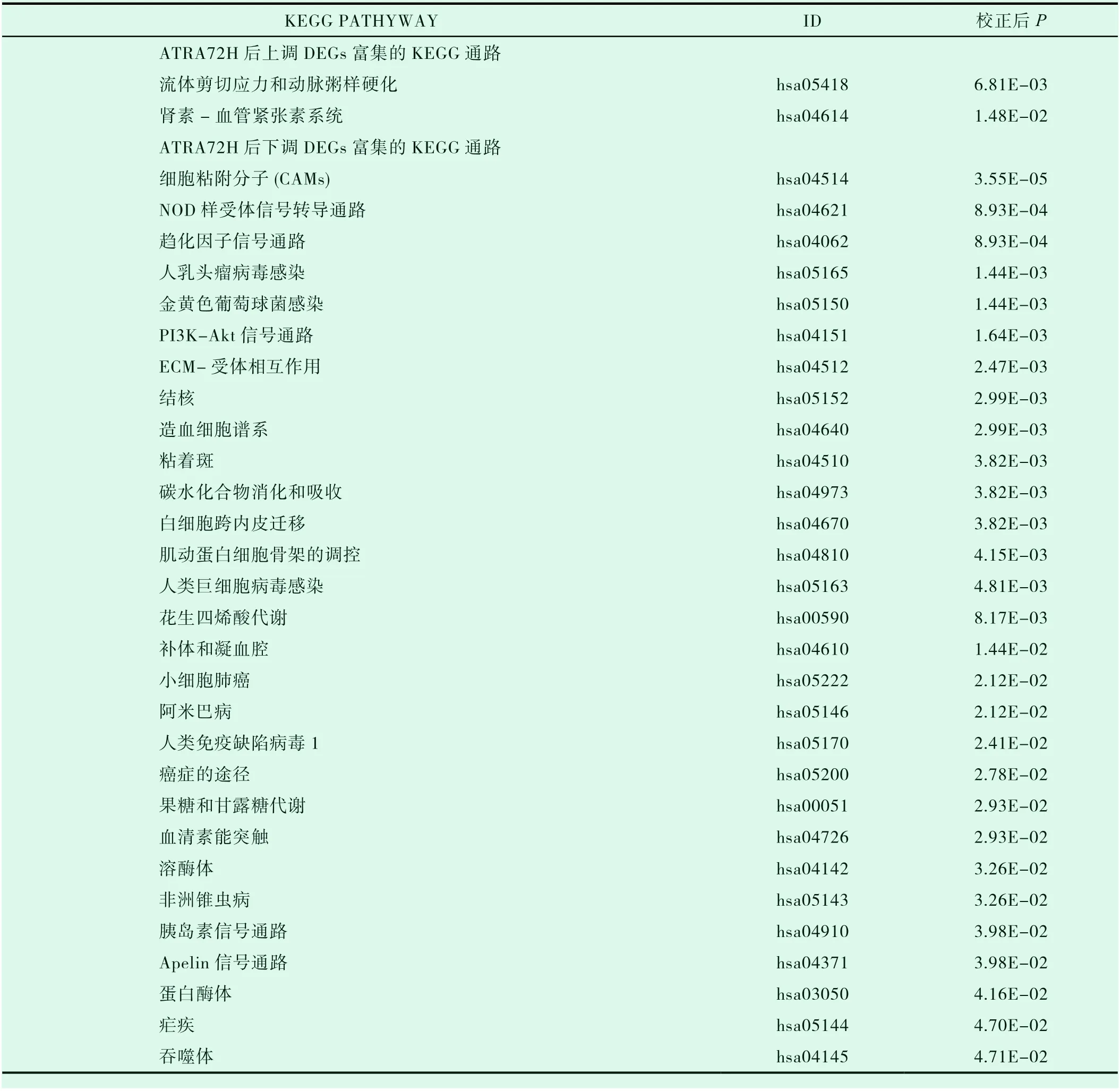
表4 UNTREATED vs ATRA72H 上、下調DEGs通過KEGG分析的主要通路

圖9 UNTREATED vs ATRA48H 上調DEGs通過GO富集的主要通路
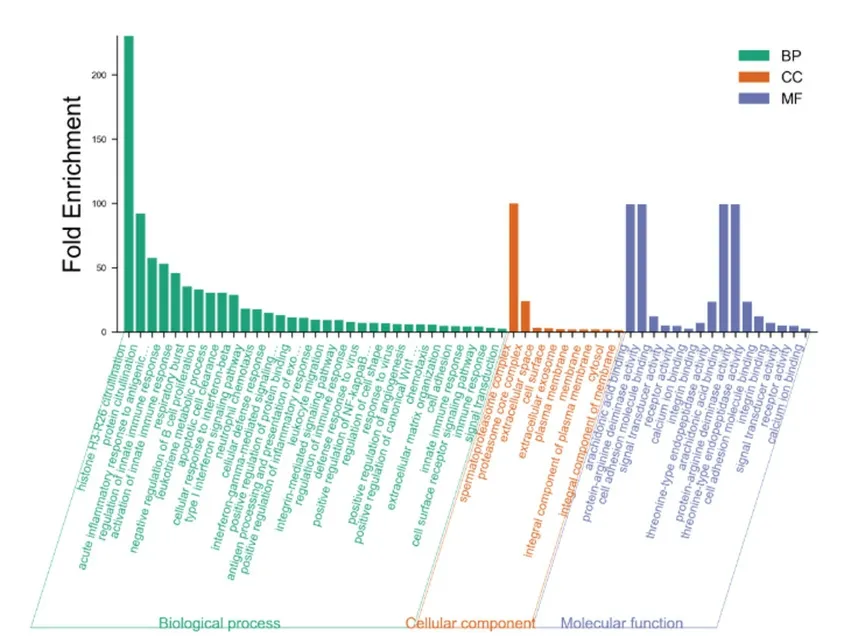
圖10 UNTREATED vs ATRA48H 下調DEGs通過GO富集的主要通路

圖11 UNTREATED vs ATRA72H 上調DEGs通過GO富集的主要通路
3.4 PPI網絡構建及分析
通過 STRING 數據庫構建 PPI網絡(圖13和14)以及Cytoscape軟件中的CytoHubba插件進一步明確ATRA48H后上、下調DEGs所涉及的蛋白相互作用及其中參與調控的關鍵基因(圖15和16),結果顯示,上調DEGs的PPI網絡中包括148個節點蛋白的145種關系,下調DEGs的PPI網絡中包括115個節點蛋白的241種關系,其中多數蛋白間都存在相互作用關系,篩選出下調DEGs排名前十的關鍵基因有:PSMB8、CXCR2、GNG2、CXCL3、PSMB9、CXCL6、P2RY13、TAS2R10、TAS2R7、PSMA3。用同樣方法得出ATRA72H后上、下調DEGs所涉及的蛋白相互作用圖(圖17和18)及其中參與調控的關鍵基因(圖19和20),結果顯示,上調DEGs的PPI網絡中包括205個節點蛋白的261種關系,下調DEGs的PPI網絡中包括166個節點蛋白的508種關系,其中多數蛋白間都存在相互作用關系,篩選出下調DEGs排名前十的關 鍵 基 因 有:ITGAM、ITGB2、ITGAX、IRF1、OAS1、OAS3、IFI35、LILRB2、IRF2、IFI6。
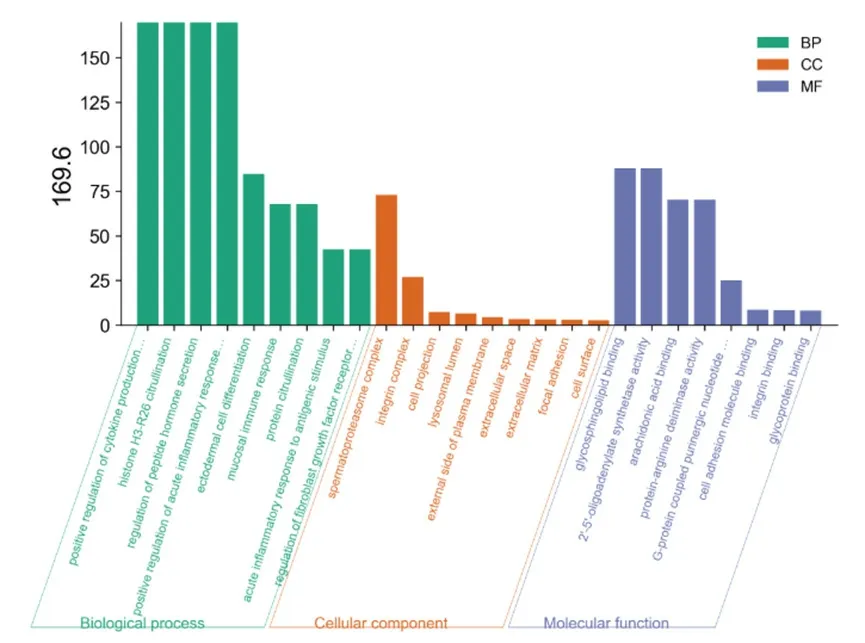
圖12 UNTREATED vs ATRA72H 下調DEGs通過GO富集的主要通路
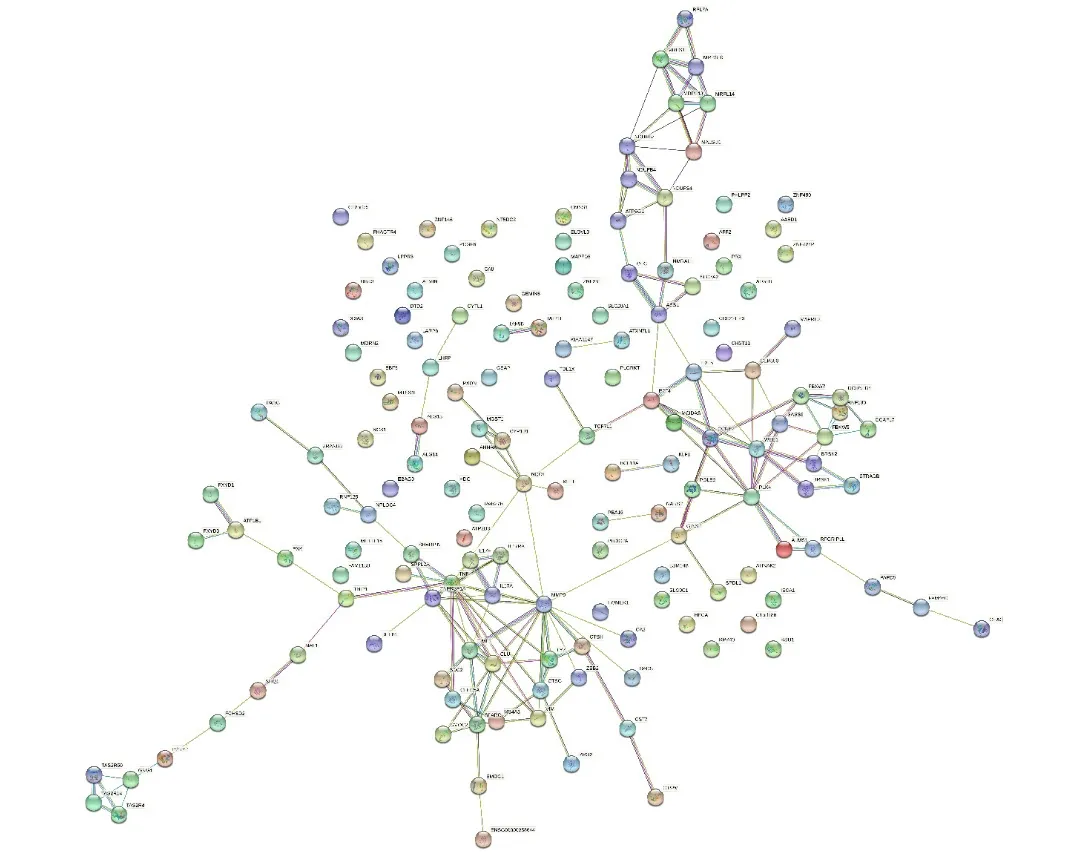
圖13 UNTREATED vs ATRA48H 上調DEGs的PPI網絡圖
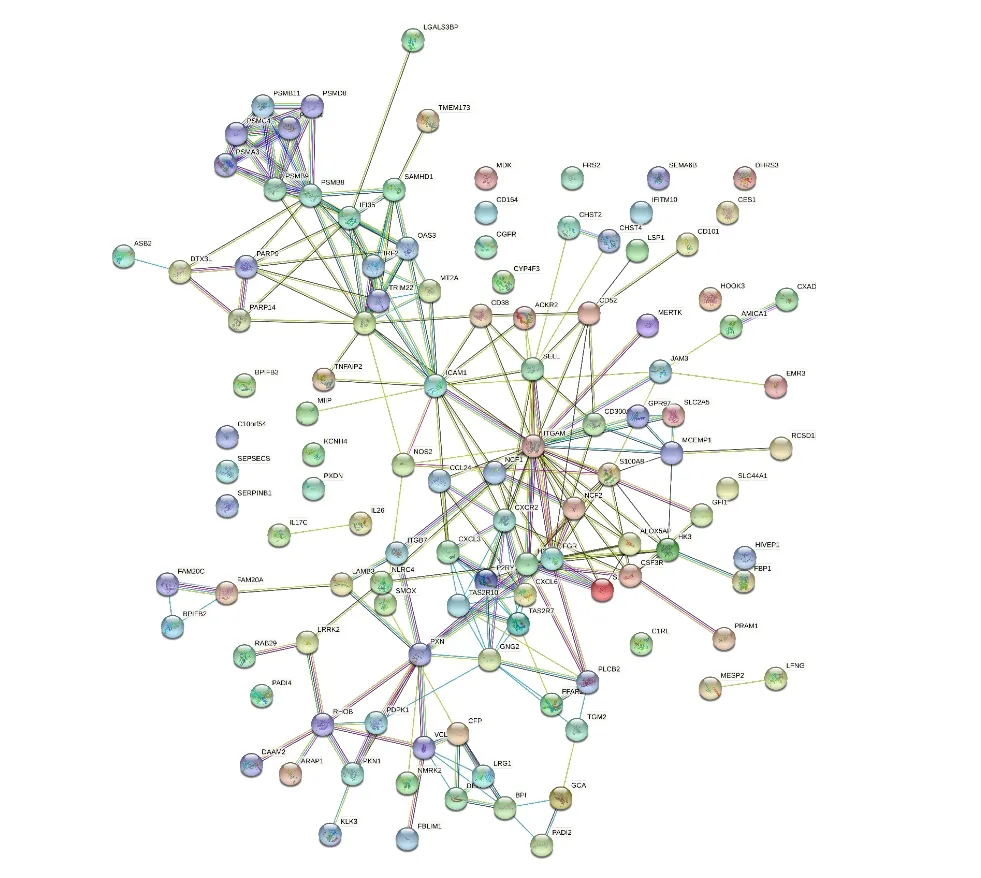
圖14 UNTREATED vs ATRA48H 下調DEGs的PPI網絡圖

圖15 UNTREATED vs ATRA48H 上調DEGs的前10個關鍵基因

圖16 UNTREATED vs ATRA48H 下調DEGs的前10個關鍵基因

圖17 UNTREATED vs ATRA72H 上調DEGs的PPI網絡圖

圖18 UNTREATED vs ATRA72H 下調DEGs的PPI網絡圖

圖19 UNTREATED vs ATRA72H 上調DEGs的前10個關鍵基因
4 討論
本文分析了ATRA處理NB4細胞后的差異基因,GO分析結果表明上調DEGs主要與一氧化氮的生物合成過程、細胞周期調控、葡萄糖進入細胞的負調控、異源代謝過程、RNA聚合酶II轉錄預起始復合物裝配、建立平面極性、宿主共生體生長的負調控;而下調的DEGs主要與炎癥反應、中性粒細胞趨化途徑、細胞粘附、細胞凋亡、免疫反應、蛋白質合成相關。同時在 KEGG 分析發現差異基因主要富集在趨化因子信號通路、細胞粘附分子、NOD樣受體信號轉導通路、白細胞跨內皮遷移、PI3K-Akt信號傳導途徑。另外,在DEGs形成的PPI網絡進行分析,可觀察到下調基因參與的通路更加具有相關性,設計的通路和關系節點更加密集,對ATRA48H和ATRA72H后下調DEGs的PPI網絡中分別篩選出前10 位與ATRA處理后NB4細胞表達下調的關鍵基因分別繪制生存曲線(組圖23),發現6個高表達提示預后不良,包括:OAS1、OAS3、PSMB9、PSMB8、ITGAM、IFI35可作為AML的危險因素,為AML的治療提供了新靶點。

圖20 UNTREATED vs ATRA72H 下調DEGs的前10個關鍵基因

圖21 KEGG趨化因子通路,紅色代表上調DEGs,藍色代表下調DEGs,

圖22 KEGG白細胞跨內皮遷移通路,紅色代表上調DEGs,藍色代表下調DEGs

圖23 ATRA處理后NB4細胞表達下調的關鍵基因在AML中的生存曲線
猜你喜歡
中學生數理化(高中版.高考理化)(2021年6期)2021-07-28 06:21:04
民用飛機設計與研究(2020年4期)2021-01-21 09:15:02
人大建設(2019年9期)2019-12-27 09:06:30
電子制作(2018年18期)2018-11-14 01:48:24
山東工業技術(2016年15期)2016-12-01 05:31:22
中國中醫藥現代遠程教育(2014年11期)2014-08-08 13:23:44
NBA特刊(2014年7期)2014-04-29 00:44:03
終身教育研究(2014年5期)2014-02-28 01:23:06
中國商人(2013年1期)2013-12-04 08:52:52
兒童時代(2009年5期)2009-05-21 05:31:26
