不同改性方法對花生蛋白理化特性影響研究
2021-11-12 00:55:46張煥麗郭世閔鐘熳肖志剛
中國糧油學報 2021年9期
關鍵詞:改性
李 響 張煥麗 郭世 陳 嵐 閔鐘熳 肖志剛
(沈陽師范大學糧食學院1,沈陽 110034)(華南理工大學食品科學與工程學院2,廣州 510641)
花生是我國重要的糧食作物,種植范圍廣、產量高,富含脂肪、蛋白質等多種營養(yǎng)物質,是全球第三位的植物蛋白質來源[1]。隨著我國經(jīng)濟水平的發(fā)展和生活質量的提高,花生類產品的需求量也逐步上升[2]。由于花生中各類營養(yǎng)素比較全面且營養(yǎng)價值均衡,因此被認為是理想的高蛋白、高脂肪優(yōu)質健康食品[3]。與其他植物源蛋白相比,花生蛋白具有較高的消化性,其有效利用率高達98%,易于人體的消化吸收[4],其中賴氨酸有效利用率達到98.8%,而大豆蛋白中的有效利用率僅為78%[5],比大豆的高出20%。研究表明,花生中含有的抗營養(yǎng)因子要明顯少于大豆,因此,花生可替代蛋白基料和牛乳等動物奶類為乳糖不耐癥人群提供適宜的高蛋白食品[6]。
目前,許多專家學者開始對花生蛋白特性進行研究,為了進一步改善花生蛋白的特性,可對花生蛋白進行改性處理,常用的改性方法有物理法、化學法和酶法等,現(xiàn)有改性方法中也會采用復合法對花生蛋白進行改性處理。He等[7]研究了高壓對花生分離蛋白的理化特性的影響,高壓處理后發(fā)現(xiàn),花生分離蛋白的持水力、持油率以及油結合能力均顯著改善。熊柳[8]對花生分離蛋白進行磷酸化改性,改性后花生分離蛋白的功能特性(如吸油性、吸水性、乳化性、持水性、乳化穩(wěn)定性、泡沫穩(wěn)定性)均不同程度的提高。張兆麗研究通過酶法處理花生分離蛋白,得出堿性蛋白酶改性的最佳條件,改性后,花生分離蛋白的吸水性、乳化性均得到顯著改善。Dong等[9]將高壓均質聯(lián)合酶處理技術對花生蛋白進行復合改性,提高了花生蛋白的酶解程度,產物中的小分子肽含量明顯增多。現(xiàn)有研究結果表明,單一改性方法對花生蛋白改性的效率不高[10],鮮有復合法對花生蛋白進行改性的研究。擠壓膨化改性法利用高溫、高壓和高剪切作用,可顯著破壞花生蛋白結構,暴露更多的酶切位點[11,12],反應效率高,對花生蛋白影響較顯著。酶法改性因其條件溫和,專一性較高,是對花生蛋白改性有效方法之一。復合改性具有改性效果好、成本低等優(yōu)點, 因此, 在實際生產中的應用越來越廣泛。
本實驗采用擠壓、酶法和擠壓協(xié)同酶解3種方法對花生蛋白進行改性處理,通過溶解性、乳化性、乳化穩(wěn)定性、起泡性等方法對花生蛋白的功能特性進行分析,采用傅里葉變換紅外光譜(Fourier transform infrared spectroscopy,F(xiàn)T-IR)、圓二色譜(circular dichroism,CD)和聚丙烯酰胺凝膠電泳(polyacrylamide gel electrophoresis,SDS-PAGE)、蛋白質水解度等現(xiàn)代分析手段,研究花生蛋白結構、分子質量等的變化,進一步探討3種改性方法花生蛋白理化性質的影響,結合蛋白空間結構模型,以期為開發(fā)花生蛋白適宜營養(yǎng)產品,提高花生蛋白類食品的附加價值提供參考。
1 材料與方法
1.1 材料與試劑
花生蛋白(PPI)、牛血清蛋白、堿性蛋白酶、十二烷基硫酸鈉(SDS)、丙烯酰胺(Acr)、三羥甲基氨基甲烷(Tris)、甘氨酸、過硫酸銨、四甲基乙二胺(TEMED)、考馬斯亮藍R-250、溴酚藍、二硫蘇糖醇(DTT)、蛋白樣標準品(電泳純)、冰乙酸、甲醇、β-巰基乙醇、磷酸二氫鈉。
1.2 儀器與設備
DWF-100電動粉碎機,HH-4Nicolet Nexus 470傅里葉紅外光譜儀,Scientz-12 N冷凍干燥機,SDS-PAGE電泳儀,DS56-Ⅲ雙螺桿擠壓膨化機,DELTA 320 pH計,UV-1200S紫外可見光分光光度計,J-1100圓二色譜儀。
1.3 方法
1.3.1 花生蛋白的改性
擠壓膨化:以溶解性為指標, 花生蛋白擠壓改性的工藝參數(shù)經(jīng)過前期優(yōu)化實驗確定,準確稱取3.0 kg純度為92%的PPI,調整原料含水量至25%,機筒溫度為100 ℃,螺桿轉速為300 r/min,喂料速度10 kg/h。蛋白擠出物經(jīng)真空冷凍干燥后,粉碎過100目篩,放入自封袋中備用。
酶法:以溶解性為指標, 花生蛋白酶解改性的工藝參數(shù)經(jīng)過前期優(yōu)化實驗確定為采用堿性蛋白酶對未處理的PPI進行酶解。酶解條件為:溫度60 ℃、pH 8.0、底物蛋白質量濃度8%、酶活216735 U/g、酶活與底物蛋白比例為100 U/g、酶解時間60 min。將酶解處理后的樣品經(jīng)真空冷凍干燥后放入自封袋中,備用。
擠壓協(xié)同酶法:準確稱取3.0 kg擠壓預處理的PPI,利用堿性蛋白酶進行酶解,擠壓和酶解條件分別同酶法。處理后的樣品經(jīng)真空冷凍干燥后放入自封袋中,備用。
1.3.2 蛋白質水解度測定(DH)
采用OPA發(fā)法對花生蛋白水解度進行測定,用紫外分光光度計測量酶底物的DH,以精氨酸作為標準品,繪制標準曲線[13]。計算公式見式(1)。
h=(ht-hc)×DF
(1)
式中:ht為樣品的濃度;hc為對照品(未水解樣品)的濃度;htot為7.8 mmol/g;DF為樣品的稀釋倍數(shù)。
1.3.3 蛋白質溶解性的測定
參考Shimada等[14]的方法,將處理前后的花生蛋白樣品溶于超純水中,用1 mol/L的HCl和NaOH來調節(jié)pH,使其pH處于7,在水浴鍋中攪拌1 h(室溫),攪拌均勻后于室溫下離心30 min(10 000 r/min),取上清液,采用Bradford法測定上清液中蛋白含量[15],用凱氏定氮法測定樣品中總蛋白含量,根據(jù)上清液中的蛋白占樣品總蛋白含量的百分比計算溶解度,并以氮溶解指數(shù)表示(NSI%),見式(2)。
(2)
1.3.4 蛋白質乳化性及乳化穩(wěn)定性的測定
采用濁度法[16],并稍加改動:將不同處理的樣品(花生分離蛋白作對照)溶于0.1 mol/L的磷酸鹽緩沖溶液(pH 7.0)中,配成 0.1%質量濃度的樣品溶液。將8 mL大豆油加入30 mL樣品溶液中,然后用高速勻質機在室溫條件下均質2 min(13 500r/min)。分別在均質結束后和靜置10 min后立即從底部吸取100 μL的樣品,然后用10 mL 0.1%的 SDS(m)進行稀釋,測定3次500 nm處吸光度值,分別記為A0和A10,乳化活性指數(shù)(Emulsification Activity Index,EAI)按式(3)計算。
(3)
式中:EAI為乳化活性指數(shù)/m2/g;N為稀釋倍數(shù);φ為體系中油相占比;C為蛋白質量濃度/g/mL;L為比色池光徑/1 cm。
乳化穩(wěn)定性(Emulsification Stability Index,ESI)按式(4)計算。
(4)
式中:ESI為乳化穩(wěn)定性指數(shù)/min;A0為0 min時的吸光度值;ΔT為測定乳化性的兩次時間間隔,本實驗取10 min;A10為10 min時的吸光度值。
1.3.5 蛋白質起泡性及泡沫穩(wěn)定性的測定
參考Sathe等[17]的實驗方法并稍加修改,首先配制100 mL花生分離蛋白樣品溶液(1%,pH 7.0),置于500 mL量筒中,然后于室溫條件下用高速勻質機在17 500 r/min條件下連續(xù)均質3次,每次40 s。起泡能力(Foaming Capacity)和泡沫穩(wěn)定性(Foaming Stability)分別按式(5)和式(6)計算。
(5)
(6)
式中:FC為起泡能力/%;FS為泡沫穩(wěn)定性/%;V0為均質后的液面高度;V30為靜置30 min后再次記錄液面高度。
1.3.6 傅里葉變換紅外光譜法(FT-IR)
將樣品與溴化鉀混合,置于研缽中研細混合均勻,采用壓片法制樣,將其置于紅外光譜分析儀進行測試,掃描波數(shù)范圍為 4 000~400 cm-1,分辨率為4 cm-1。
1.3.7 圓二色光譜(CD)
準確稱取0.2 mg/mL蛋白質溶液,將其置于190~250 nm波長范圍內,以100 nm/min的速率掃描,室溫下帶寬為1 nm,路徑長度為0.1 cm。使用磷酸鈉緩沖液(10 mol/L,pH 7.0)校正基線。并用Jasco二級結構軟件計算不同二級結構的含量。
1.3.8 十二烷基硫酸鈉-聚丙烯酰胺凝膠電泳(SDS-PAGE)
參考Backman等[18]方法并稍作修改。將樣品溶于1 mL的超純水中,取40 μL Laemmli緩沖液(0.1 mol/L Tris-HCL,pH 6.8,10%十二烷基硫酸鈉,20%甘油,溴酚藍),向混合好溶液中加入50 μL DTT,在沸水浴中加熱5 min。冷卻至室溫后,進樣后將凝膠置于恒定電流下電泳,在5%質量濃度的濃縮膠中電泳20 min,在12%質量濃度的分離膠中電泳40 min,待條帶跑至凝膠底端時,結束電泳,凝膠用甲醇溶液固定30 min,染色40 min,用冰醋酸溶液浸泡3~4次后,直到蛋白條帶清晰,將脫色后的凝膠置于凝膠成像系統(tǒng)中進行成像處理。
1.3.9 巰基(-SH)和二硫鍵(-S-S-)含量的測定。
根據(jù)Song等[19]的方法并稍作修改。將4 mg 5,5′-二硫雙歧桿菌(2-硝基苯甲酸)(DTNB)試劑溶于1 mL三甘氨酸緩沖液(pH 8.0)中制備Ellman試劑。分別稱取75 mg不同改性條件下的花生蛋白樣品快速溶于1.0 mL三甘氨酸緩沖液中,然后加入50 μL Ellman試劑,在渦旋振蕩器中振蕩混勻后在水浴鍋中25 ℃保溫15 min,用紫外可見分光光度計在412 nm處測定上清液的吸光度,用試劑緩沖液作空白對照。用摩爾消光系數(shù)13 600除以吸光度值,得到巰基含量,表示為μmol/g蛋白質。SH含量計算見式(7)。
(7)
式中:A412是樣品在412 nm處的吸光度;C為樣品質量濃度/mg/mL。
參考Benjaknl等[20]的方法并稍作改動,分別稱取一定量的不同處理條件下的花生蛋白溶解于0.6 mol/L,pH 7.5的KCl緩沖溶液中,將其稀釋至100 mg/mL,室溫下以4 000 r/min離心20 min,取0.5 mL上清液,加入3.0 mol/L,pH 9.5的NTSB檢測液,混合均勻后在室溫條件下置于暗處25 min,利用分光光度計于412 nm處測定其吸光值,二硫鍵含量以mol/106 g蛋白質計。其中摩爾消光系數(shù)(ε)為13 900/L/(mol·cm),二硫鍵含量按式(8)計算。
(8)
式中:C為二硫鍵濃度;A412為412 nm處的吸光值;D為稀釋倍數(shù)。
1.3.10 蛋白質表面疏水性(H0)的測定
采用Shi等[21]方法并稍作修改。稱取一定量的不同改性條件下的花生蛋白,并溶解于0.1 mol/L、pH 7.0的磷酸鹽緩沖液中,在20 ℃處理2 min后,將混合物在10 000 r/min離心20 min,離心結束后立即取上清液,并測定上清液的濃度,測定濃度后用磷酸鹽緩沖液稀釋,質量濃度稀釋范圍為0.05~0.4mg/mL,然后將20 μL的1-苯胺基-8-苯磺酸(ANS)添加到4 mL不同改性處理的蛋白質樣品溶液中。測試條件為:激發(fā)波長390 nm,發(fā)射波長470 nm,掃描速度為10 nm/min。熒光強度與蛋白質濃度曲線的初始斜率作為H0的指標。
1.3.11 統(tǒng)計分析
每組實驗做3次平行,數(shù)據(jù)分析采用SPSS20.0統(tǒng)計軟件,采用Duncan對數(shù)據(jù)進行顯著性分析(P<0.05),利用Origin 9.5軟件作圖。
2 結果與分析
2.1 溶解性
圖1為原花生蛋白(PPI)和擠壓膨化法處理后花生蛋白(EPPI)、酶解法處理后花生蛋白(EHPPI)、擠壓協(xié)同酶解法改性花生蛋白(EEPPI)在處理條件下的溶解性。由圖1可知,EPPI的溶解性明顯低于PPI,EHPPI和EEPPI溶解性顯著高于PPI。EPPI溶解性降低的原因可能是花生蛋白在擠壓機筒內受到高溫高壓、高剪切力作用,其分子結構發(fā)生改變,蛋白分子結構展開,部分疏水基團暴露,促進花生蛋白分子之間相互作用形成大量不溶大分子聚集體,表現(xiàn)出低于原花生蛋白的溶解性[22]。EHPPI溶解性高于PPI的原因可能是原花生蛋白具有致密的分子結構[23],而酶解過程促使花生蛋白分子的巰基斷裂,分子質量降低,肽鏈舒展,暴露出更多的親水性基團,不溶性的聚集體含量降低[24]。同時,圍繞著新暴露的極性基團的結合水逐漸增多,引起蛋白質的水化作用增強,從而表現(xiàn)出高于原花生蛋白的溶解性。從而使得溶解性增加。EEPPI在擠壓過程中蛋白由于分子熱運動破壞了束縛力,致其分子展開,二硫鍵受到破壞,原本包裹有序的狀態(tài)發(fā)生改變,特別是抗酶解的蛋白質組分,顯著增加了蛋白酶對蛋白的親和性,溶解性高于EPPI和EHPPI,綜合分析,擠壓協(xié)同酶解法效果優(yōu)于單一改性方法[25]。
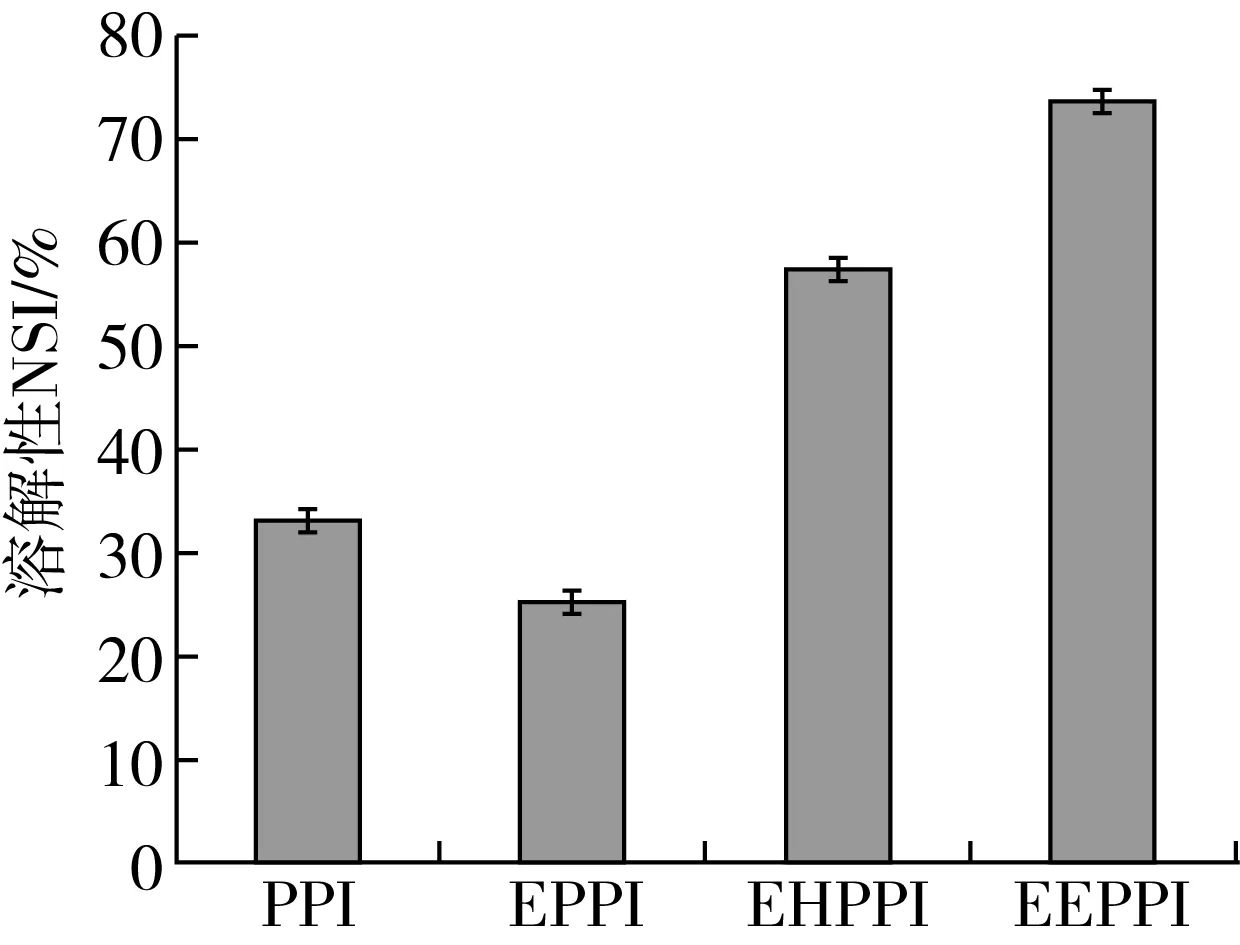
注:PPI、EPPI、EHPPI、EEPPI分別為花生蛋白、擠壓膨化法處理后花生蛋白、酶解法處理后花生蛋白、擠壓協(xié)同酶解法處理后花生蛋白。圖1 不同處理條件下花生蛋白的溶解性
2.2 蛋白質乳化性及乳化穩(wěn)定性
圖2為不同處理方式對花生蛋白乳化性和乳化穩(wěn)定性的影響。由圖2可知,與未處理花生蛋白相比,三種處理方式花生蛋白的乳化性和乳化穩(wěn)定性均得到顯著提高。擠壓處理方式提高的原因是因為擠壓膨化處理給予蛋白質短時間的高溫、高壓和高剪切作用,部分蛋白變性并發(fā)生分子結構的變化,改變蛋白質表面疏水性和親水性基團的分布位點,分子內部親油性疏水基團暴露,從而提高蛋白的乳化能力。通過比較不同處理的改性效果,EHPPI的乳化性和乳化穩(wěn)定性最高,是因為經(jīng)堿性蛋白酶水解后,花生蛋白分解成更多小分子肽段,分子內部、外部及疏水基團暴露,界面性質發(fā)生變化,溶解性增強[26],從而改善蛋白質的乳化性。同時,蛋白羧基上帶有相同電荷的液滴,相互之間存在靜電排斥作用,乳液液滴間不易于絮凝和凝聚,提高了蛋白的乳化穩(wěn)定性。EEPPI低于酶解的原因是因為擠壓協(xié)同酶解處理后蛋白的水解度較大,其分子被水解為更小分子質量的短肽,維持花生分離蛋白分子空間結構的作用力和化學鍵(包括范德華力、氫鍵、離子鍵等)被破壞程度較大,阻礙了油水界面上起到保護作用界面膜的形成,導致油滴表面的保護層變薄,小油滴逐漸聚集成大顆粒[27],從而引起乳化性和乳化穩(wěn)定性降低。

圖2 不同處理條件下花生蛋白的乳化性和乳化穩(wěn)定性
2.3 蛋白質起泡性及泡沫穩(wěn)定性
圖3為不同處理方式對花生蛋白起泡性及泡沫穩(wěn)定性的影響。由圖3可知,與原花生蛋白相比,擠壓處理顯著提高花生分離蛋白的起泡性和泡沫穩(wěn)定性,酶解和擠壓協(xié)同酶解處理只起到提高起泡性的效果,反而導致泡沫穩(wěn)定性變差。擠壓處理的花生蛋白起泡性及泡沫穩(wěn)定性提高的原因可能是經(jīng)擠壓作用,破壞了蛋白分子結構,蛋白疏水性增強,從而快速吸附至氣—液界面,界面上的蛋白質-蛋白質相互作用,形成粘稠狀的膜,同時界面和吸附分子之間也缺乏推斥力,被吸附至界面的蛋白質的數(shù)量增加,吸附在氣-水界面上的蛋白質膜的厚度和硬度的增加,疏水基團的暴露,多肽鏈的交聯(lián),界面膜層的黏度提高,增加泡沫的穩(wěn)定性[28]。酶解及擠壓協(xié)同酶解處理方式提高起泡性,降低泡沫穩(wěn)定性的原因可能是由于花生蛋白經(jīng)酶解后蛋白質分子間的肽鏈展開,疏水基團的暴露,并增加了多肽分子的界面活性,增加了界面的親和力和吸附率[29]提高了發(fā)泡的能力。由于酶解后蛋白質的黏度會變小,液膜會變得很脆弱無法裹住氣泡,使得泡沫穩(wěn)定性變差。經(jīng)擠壓處理后脫敏的花生蛋白其水解度較大,溶解性較好,起泡能力比單獨酶解處理效果強,黏度的變小,泡沫穩(wěn)定性會較差。
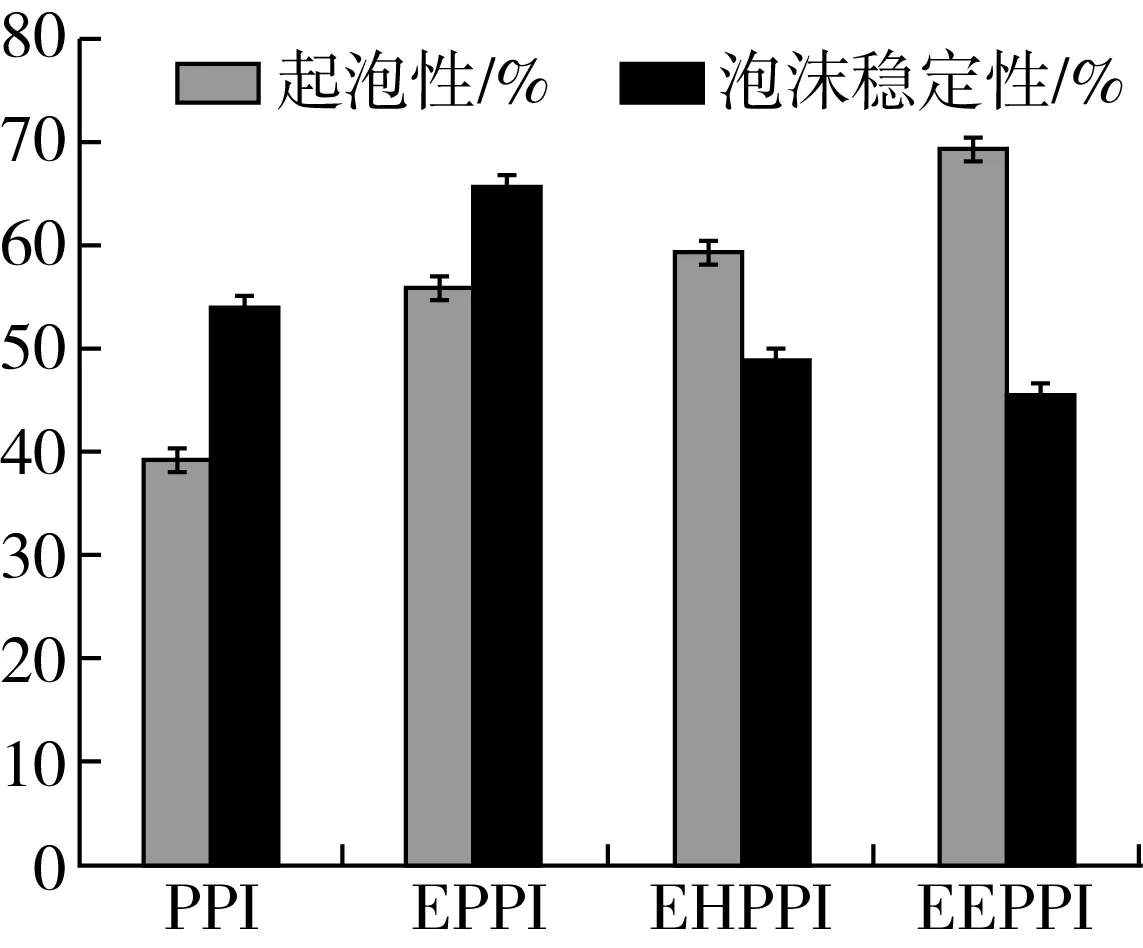
圖3 不同處理條件下花生蛋白的起泡性和泡沫穩(wěn)定性
2.4 傅里葉變換紅外光譜分析(FT-IR)
圖4為不同處理方式花生蛋白的紅外光譜圖。與PPI相比,EPPI、EHPPI和EEPPI酰胺Ⅰ譜帶的強度和波數(shù)均有明顯變化,酰胺Ⅰ譜帶振動頻率取決于羰基和氨基之間的氫鍵性質[30],可以反映多肽或蛋白質特定二級結構的變化。經(jīng)擠壓膨化法、酶法和擠壓協(xié)同酶法3種方法處理后,花生蛋白在1 621 cm-1處吸收峰強度均有不同程度的增強,表明其β-折疊含量有所增加[31]。1 652 cm-1峰寬明顯減小,表明α-螺旋含量有所減少。擠壓膨化法、酶法和擠壓協(xié)同酶法處理均會影響花生蛋白的二級結構變化。擠壓改性導致PPI的反褶積酰胺I帶峰紅移(約1 cm-1),這表明經(jīng)擠壓處理后花生蛋白分子鏈展開,EHPPI和EEPPI的紅移增加(約2 cm-1),這說明酶法和擠壓協(xié)同酶法處理有助于花生蛋白分子鏈的展開。
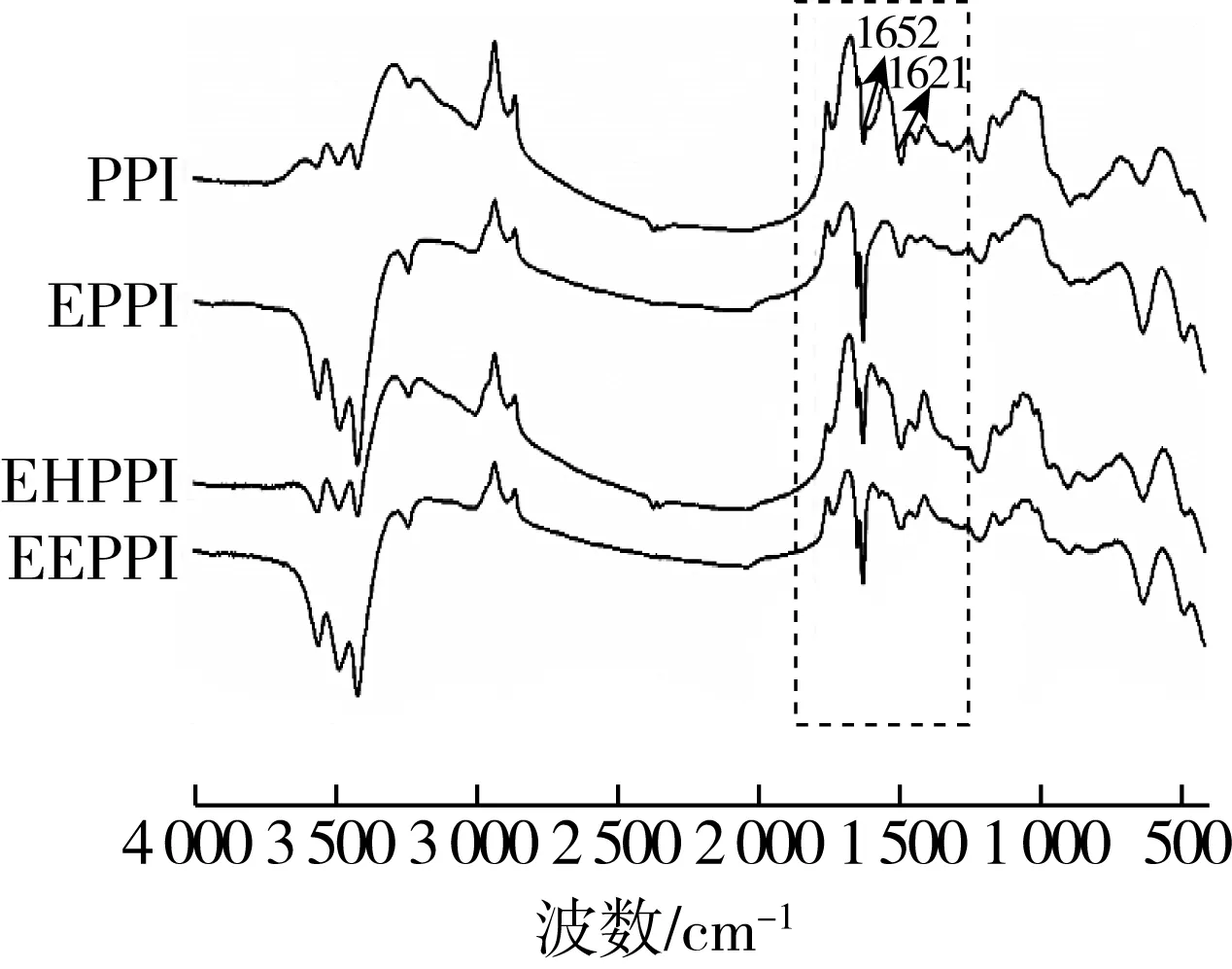
圖4 不同改性方法下花生蛋白的紅外圖譜
2.5 圓二色譜分析 (CD)
表1為花生蛋白二級結構的變化。從表1可以看出,花生蛋白經(jīng)3種方法改性后,α-螺旋和β-轉角相對含量降低,β-折疊和無規(guī)卷曲含量升高。經(jīng)擠壓膨化法處理后α-螺旋和β-轉角相對含量分別降低3.00%和2.20%,β-折疊和無規(guī)卷曲相對含量分別升高了4.10%和1.10%。酶解處理后α-螺旋和β-轉角相對含量分別降低了3.30%和3.10%,β-折疊和無規(guī)卷曲相對含量分別升高了4.60%和1.80%。擠壓協(xié)同酶法處理后蛋白質二級結構變化最明顯,α-螺旋和β-轉角含量分別降低7.30%和6.10%,β-折疊和無規(guī)卷曲含量分別升高了6.60%和6.80%。擠壓處理引起花生蛋白二級結構相對含量變化的原因可能是因為擠壓過程中產生的高溫、高剪切力破壞了蛋白質二級結構的有序性,導致部分蛋白分子變性重新聚集。酶法處理引起花生蛋白二級結構相對含量變化的原因可能是花生蛋白經(jīng)酶解處理后其花生蛋白分子鏈受到破壞,肽鏈或花生蛋白的二級結構展開,暴露出大量分子內氫鍵,形成了肽鏈間的氫鍵,導致α-螺旋含量減少,β-折疊和無規(guī)則卷曲含量增加。擠壓協(xié)同酶解處理導致花生蛋白二級結構含量相對含量變化最明顯的原因可能是,擠壓預處理破壞了蛋白質的二硫鍵,分子打開,增加了花生蛋白與酶作用的機會,有利于花生蛋白的水解。

表1 不同處理方式下花生蛋白二級結構的相對含量及水解度
2.6 巰基(-SH)和二硫鍵(-S-S-)含量分析
圖5為不同處理方式對花生蛋白巰基和二硫鍵含量的影響。如圖2所示,與PPI相比,EPPI、EHPPI和EEPPI巰基含量均有所增加,而二硫鍵含量明顯降低。EPPI、EHPPI和EEPPI的巰基分別增加了10.77%、16.27%、50.47%,二硫鍵含量分別減少了33.01%、55.28%、87.33%。這表明擠壓和酶解反應對花生分離蛋白游離巰基和二硫鍵含量都有顯著的影響。擠壓和酶解處理導致巰基含量增加的原因可能是由于蛋白質分子的高級結構增到破壞,降解,肽鏈伸展,二硫鍵部分斷裂,包裹在花生蛋白內部的巰基基團外露,巰基含量增多,二硫鍵含量減少,這與Anderson等[32]研究結果一致。
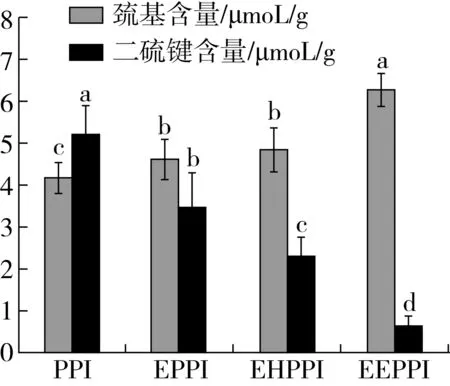
注:不同字母表示差異顯著(P<0.05),圖6同。圖5 不同處理方式下花生蛋白中巰基含量和二硫鍵含量
2.7 表面疏水性(H0)
圖6為不同處理方式對花生蛋白表面疏水性的影響規(guī)律。從圖6可以看出,PPI的表面疏水性為149.00,EPPI、EHPPI和EEPPI的表面疏水性分別為200.12、225.89、251.42,呈現(xiàn)逐漸升高的趨勢。表面疏水性的變化與蛋白質變性和熱聚集體的形成有關,說明3種處理方法改變了花生蛋白的三級結構。擠壓處理破壞了蛋白的二硫鍵,分子結構展開,改變了花生蛋白分子的空間結構。酶解處理下破壞了蛋白質的相互作用,誘導蛋白分子解折疊展開更明顯,花生蛋白被逐漸酶解為小片段多肽,包埋于蛋白質分子內部的疏水性氨基酸趨于“暴露態(tài)”,從而導致蛋白質表面疏水性增加[33]。擠壓后增加了無規(guī)則卷曲含量,此時更有利于酶解進行,使得包埋于球蛋白分子內部的疏水基團外露, ANS熒光探針結合位點增加,所以擠壓協(xié)同酶法改性能夠促使花生蛋白分子結構得到充分的伸展,H0達到最大值[34]。擠壓協(xié)同酶解處理改變蛋白質高級結構,暴露更多的酶作用位點,蛋白質表面疏水性與游離巰基含量呈線性正相關,這與臧學麗等[35]研究結果一致。
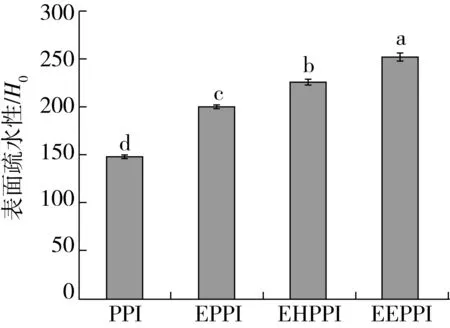
圖6 不同處理方式對花生蛋白的表面疏水性影響
2.8 花生蛋白水解程度及十二烷基硫酸鈉-聚丙烯酰胺凝膠電泳分析(SDS-PAGE)
不同處理方式下花生蛋白SDS-PAGE的影響如圖7所示。各個處理組在EPPI凝膠電泳圖上沒有看到明顯的條帶變化,EHPPI和EEPPI凝膠電泳圖上有明顯的條帶變化,這表明在擠壓膨化處理方式可以改變花生蛋白的二、三、四級結構,而一級結構不受溫度和機械力影響。酶解后的花生蛋白泳道中分子質量為17~20、36、38、43、64 ku均有不同程度變淺,在14.4 ku處有彌散性條帶存在,說明堿性蛋白酶已經(jīng)能夠將花生蛋白水解至14.4 ku以下的小分子蛋白質片段。擠壓膨化協(xié)同酶解處理后堿性蛋白酶花生蛋白泳道中分子質量為64、43、38、36 ku條帶徹底消失,17~20 ku條帶明顯變淺,在14.4 ku處也有彌散性條帶存在,這可能是擠壓膨化處理使二、三、四級緊密結構展開[36],暴露更多的酶解位點,使酶分子更容易進入蛋白質內部,促進了堿性蛋白酶的水解,小分子蛋白片段增多。從14.4 ku處條帶深淺來看,酶解和擠壓協(xié)同酶解處理小分子蛋白片段并沒有明顯的區(qū)別,說明SDS-PAGE圖不能判斷擠壓協(xié)同酶解處理花生蛋白是否能得到更低分子質量的蛋白片段[37]。

圖7 不同處理方式下花生蛋白的凝膠電泳圖
3 結論
實驗選擇對花生蛋白的理化性質進行了研究。研究結果表明,擠壓、酶解、擠壓協(xié)同酶法3種改性方式均為有效的花生蛋白改性手段,綜合對比下擠壓協(xié)同酶解法對花生蛋白理化性質影響更為明顯。對3種改性花生蛋白的功能特性進行分析,其溶解性、乳化性以及起泡性均有所提高,通過紅外光譜、電泳及圓二色譜結果分析發(fā)現(xiàn),蛋白質二級結構被破壞,平均分子質量降低,α-螺旋結構比例大幅下降、β-折疊和自由卷曲結構比例的提升,巰基含量增加,二硫鍵含量降低,表面疏水性指數(shù)增大。擠壓協(xié)同酶法改性花生蛋白,對實際生產中產品的質量必然起到積極作用,可提高花生蛋白產品的附加價值,增加其產品的應用范圍。
猜你喜歡
紡織科學研究(2020年1期)2020-05-21 00:31:06
中國塑料(2016年12期)2016-06-15 20:30:07
中國塑料(2016年2期)2016-06-15 20:30:00
中國塑料(2016年2期)2016-06-15 20:29:59
中國塑料(2016年5期)2016-04-16 05:25:36
廣西林業(yè)科學(2016年3期)2016-03-16 05:43:30
中國塑料(2015年3期)2015-11-27 03:41:38
中國塑料(2015年11期)2015-10-14 01:14:14
中國塑料(2015年9期)2015-10-14 01:12:17
中國塑料(2015年4期)2015-10-14 01:09:19
