Gitelman綜合征、Bartter綜合征各1例并文獻復(fù)習(xí)
2021-11-18 09:00:13劉敏文香廖莉
中國藥學(xué)藥品知識倉庫 2021年8期
劉敏 文香 廖莉

摘要:目的:分析1例Gitelman綜合征及1例Bartter綜合征患兒的臨床表現(xiàn)及基因檢測結(jié)果,以提高臨床醫(yī)師對該類疾病的認識。方法:分析我院收治的1例Gitelman綜合征及1例Bartter綜合征患兒的臨床資料,并結(jié)合文獻資料進行分析。結(jié)果:兩例患兒臨床上均表現(xiàn)為頑固性低血鉀、低氯性代謝性堿中毒、血壓正常,其中GS患兒伴有低鎂血癥。Gitelman綜合征患兒基因檢測提示SLC12A3基因有2個雜合突變,突變位置為chr16-56906567(核苷酸變化c.965-1G>A)和chr16-56902264(核苷酸變化c.485delT)。Bartter綜合征患兒基因檢測提示CLCNKB基因突變,突變位置chr1-6381956(核苷酸變化c.1783C>T).結(jié)論:提高對該類疾病的認識,以防漏診、誤診。
關(guān)鍵詞: Gitelman綜合征;Bartter綜合征;診斷及治療
[Abstract] OBJECTIVE: To analyze the clinical manifestations and genetic test results of 1 child with Gitelman syndrome and 1 child with Bartter syndrome, so as to improve the clinician's understanding of the disease. Methods: The clinical data of 1 case of Gitelman syndrome and 1 case of Bartter syndrome in our hospital were analyzed, and related literatures were reviewed.Results: The clinical manifestations of both children were refractory hypokalemia, hypochlorite metabolic alkalosis, and normal blood pressure. Hypomagnesemia was associated with GS children. Genetic testing of children with Gitelman syndrome revealed that the SLC12A3 gene has two heterozygous mutations at the positions of chr16-56906567 (nucleotide change c.965-1G> A) and chr16-56902264 (nucleotide change c.485delT). Genetic testing of children with Bartter syndrome revealed a mutation in the CLCNKB gene and the mutation location chr1-6381956 (nucleotide change c.1783C> T). Conclusion: Improve the understanding of this disease to prevent misdiagnosis and misdiagnosis.
[Key words] Gitelman syndrome;Bartter syndrome;diagnosis and therapy
【中圖分類號】G644.5 ? ? ? ? ? ? 【文獻標識碼】A ? ? ? ? ? ? 【文章編號】2107-2306(2021)07--03
Gitelman綜合征(Gitelman syndrome,GS) 和Bartter綜合征(Bartter syndrome,BS)是由不同基因突變引起的常染色體遺傳性失鹽性腎小管疾病,臨床上均表現(xiàn)為低血鉀、低氯性代謝性堿中毒、高腎素-血管緊張素-醛固酮,而血壓正常。低鎂血癥、低尿鈣在臨床上常見于GS患者。該類疾病發(fā)病率較低并且臨床醫(yī)生認識不足,主要靠基因檢測確診。本研究總結(jié)了我科收治的1例GS和1例BS患兒的臨床資料,并參閱相關(guān)文獻對兩種疾病的臨床特征、實驗室檢查、基因診斷、治療及隨訪等方面進行分析,為臨床醫(yī)務(wù)工作者提供診治思路,以便早診斷、早治療,緩解患兒癥狀,減少并發(fā)癥。
1 ?臨床資料
1.1 Gitelman綜合征1例
患兒男,8歲11月,因“嘔吐、腹瀉1天”于2015年11月10日~2015年11月25日我科住院治療。該患兒自幼易跌倒,5歲時因“鼻竇炎”于重醫(yī)兒童醫(yī)院住院治療5天,住院期間曾測血清鉀離子<3 mmol/L,當時并未明確低鉀原因。此次患兒因消化道癥狀入院,病程中共嘔吐6次,腹瀉2次,伴手足麻木感;既往無口干、多尿、多飲史,無腹痛、腹瀉病史,否認利尿劑及其他藥物使用史,飲食及睡眠可;家中無類似疾病史。
患兒身高120 cm,體重20 kg(均小于2SD),體質(zhì)量指數(shù)13.9 kg/m2,血壓90/58 mmHg,心、肺、腹部無異常。住院期間多次血鉀波動在2.48~3.31 mmol/L,其中血鉀為3.11 mmol/L時對應(yīng)的24小時尿鉀為28 mmol;血氯為92.20~102 mmol/L;血鎂為0.46~0.53 mmol/L;多次血鈉、血鈣、血磷均正常。11月24日血氣示:PH 7.461,PaO2 84 mmHg,PaCO2 34 mmHg,HCO3- 24.8 mmol/L,BE 0.4 mmol/L.大便常規(guī)及培養(yǎng)、血糖、肝腎功、甲功五項均正常;ECG,肝、膽、胰、脾、雙腎、腎上腺B超均正常;骨密度檢測:骨齡﹤5歲,骨齡攝片正常。基因檢測結(jié)果回示:SLC12A3基因有2個雜合突變,突變位置為chr16-56906567(核苷酸變化c.965-1G>A)和chr16-56902264(核苷酸變化c.485delT),分別來自父親與母親,其中一個位點為剪切突變,另一個位點為移碼突變。
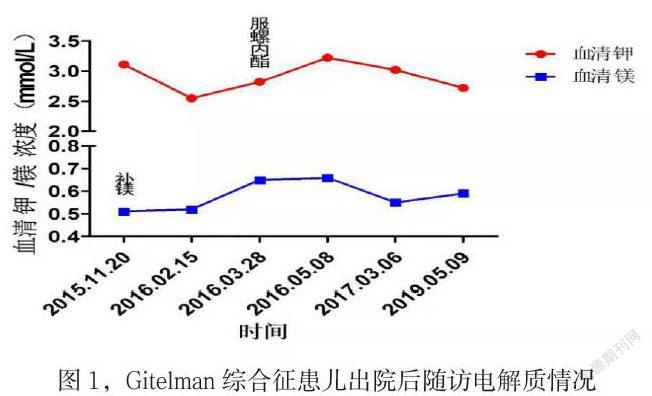
該患兒存在低血鉀、低血氯、代謝性堿中毒、低血鎂、血壓正常,基因結(jié)果提示SLC12A3基因突變,臨床診斷“Gitelman綜合征”,并給予規(guī)范補鉀、補鎂治療,隨訪電解質(zhì)情況如下圖:該患兒在補鉀基礎(chǔ)上增加螺內(nèi)酯后隨訪血鉀能維持在3.0 mmol/L以上,正規(guī)補鎂后血鎂能維持在0.6 mmol/L以上,但后期未規(guī)律服藥及隨訪,導(dǎo)致電解質(zhì)維持欠佳。
1.2 Bartter綜合征1例
患兒男,1歲11月,因“咳嗽1周”入院。患兒生后1+月生長發(fā)育逐漸落后,平素飲食差,有便秘,無煩渴、夜尿增多、頻繁嘔吐、抽搐等表現(xiàn)。8+月時因“支氣管肺炎、輪狀病毒性腸炎、重度營養(yǎng)不良”于我科住院治療,期間查電解質(zhì)發(fā)現(xiàn)血鉀2.74 mmol/L,經(jīng)補鉀治療1周,復(fù)查血鉀3.0 mmol/L后要求出院,出院后未規(guī)律隨訪。
患兒身高77 cm,體重9 kg(均小于2SD),體質(zhì)量指數(shù)15.2 kg/m2,血壓70/54 mmHg,雙肺呼吸音粗,可聞及濕羅音,心、腹部無異常。入院后查血鉀波動在2.17-2.73 mmol/L,尿鉀28.27 mmol/24h,PH7.458-7.486,HCO3- ?27.28-29.29 mmol/L.血氯 90 mmol/L.兒童醫(yī)院查腎素仰臥位測定>500 uIU/ml,醛固酮仰臥位測定55.8 ng/dl。腎功、甲功基本正常,腎臟、甲狀腺、腎上腺彩超無異常。基因結(jié)果提示chr1-6381956(核苷酸變化c.1783C>T).
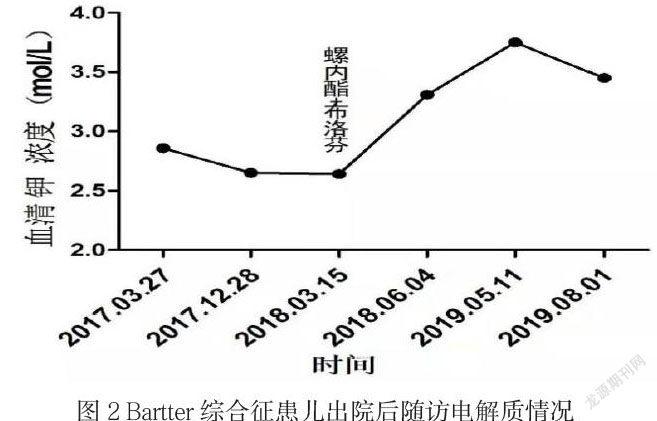
該例患兒系幼兒,嬰兒期起病,有生長發(fā)育遲緩,血壓正常,代謝性堿中毒,低鉀血癥,伴低氯血癥,血鈣、血鎂正常,高腎素-醛固酮血癥,基因結(jié)果提示CLCNKB基因突變,臨床診斷Bartter綜合征III型,并予規(guī)范靜脈及口服補鉀治療,出院后隨訪血鉀情況如下
2 ?討論:
Bartter 綜合征和Gitelman 綜合征是由 Frederic Bartter 和 Gitelman 分別于1962 年和 1966 年提出的臨床表現(xiàn)相似的失鹽性腎小管疾病[1-2],兩者均是常染色體遺傳性疾病,其共同特點為低血鉀、低氯性代謝性堿中毒、RASS系統(tǒng)激活、血壓正常或偏低,腎組織病理學(xué)檢查提示腎小球旁器增生和肥大。BS與 GS 臨床表現(xiàn)類似,不同點在于GS同時伴有低血鎂、低尿鈣,這對臨床醫(yī)生正確鑒別BS、GS尤為重要。GS的確切發(fā)病率目前尚不清楚,國外報道歐洲人中約為1/40000,亞洲人可能更高,日本人中根據(jù)雜合子攜帶率估算的患病率在10.3/10000[3,4],該病一般在青少年或成年時發(fā)病,血尿生化異常可早于臨床癥狀出現(xiàn),多數(shù)患者臨床表現(xiàn)為疲勞、乏力、多尿、口渴等非特異性癥狀,大部分患者經(jīng)治療后預(yù)后良好,但也有少數(shù)患者可出現(xiàn)生長發(fā)育遲緩、橫紋肌溶解、軟骨鈣化、室性心律失常等嚴重臨床癥狀[5],同時長期的低鉀低鎂可能引起腎功能受損、糖代謝異常、甲狀旁腺功能低下等并發(fā)癥[6-7] 。BS的發(fā)病率為1/1000000,較GS少很多,其可能因為一部分該病患者尚未確診即導(dǎo)致宮內(nèi)或新生兒死亡[8]。可發(fā)生于任何年齡段,其中兒童多見,大多數(shù)在5歲前起病,女性多于男性,嬰幼兒期發(fā)病者多有智力低下、生長發(fā)育遲緩等表現(xiàn);成人病例發(fā)病年齡多見于20歲以后,臨床上可表現(xiàn)為多尿、消瘦、手足抽搐、嘔吐、乏力等,起病隱匿,癥狀相對較輕。
Bartter 綜合征的診斷目前國內(nèi)外尚無統(tǒng)一標準,《臨床兒科學(xué)》提出BS的診斷要點如下[9]:(1)低血鉀;(2)高尿鉀:血K+<3.5 mmol/L時,尿K+>25 mmol/L;血K+<3.0 mmol/L時,尿K+>20 mmol/L; (3)代謝性堿中毒;(4)血漿腎素—血管緊張素—血漿醛固酮水平明顯增高;(5)血壓正常,且對血管加壓素(AVP) 和血管緊張素 II(AT-2)無血壓升高反應(yīng);(6)血漿前列素增高;(7)腎活檢病理學(xué)檢查提示腎小球旁器顆粒細胞明顯增生和肥大。需排除腎功能不全,繼發(fā)因素如甲亢、反復(fù)嘔吐、濫用利尿劑及緩瀉藥物等導(dǎo)致的假性BS。
2017 KDIGO指南上[10]Gitelman 綜合征診斷標準為:在 Bartter 綜合征的診斷基礎(chǔ)上加上低血鎂(Mg+<0.7 mmo1/L),低尿鈣(尿鈣/尿肌酐<0.2 或 0.1),氯排泄分數(shù)>0.5%。基因診斷為 Bartter、Gitelman 的金標準。
根據(jù)分子遺傳學(xué)角度將BS分為I-V型,如下圖。I和II型臨床表現(xiàn)相似,通常較嚴重,可導(dǎo)致妊娠期羊水過多和早產(chǎn)。嬰兒期存活下來的患者可發(fā)生低鉀血癥、代謝性堿中毒、多尿癥和高鈣尿癥。II型是位于染色體 11 q24-25 上的 KCNJ1 基因突變。其編碼為造成 ATP敏感的腎臟內(nèi)向調(diào)節(jié)腎小管 K+通道 ROMK 功能失活,存在該突變的新生兒通常最初發(fā)生高鉀血癥[11],隨著患兒成長,其他鉀通道會發(fā)揮活性,促使發(fā)生低鉀血癥。上述患者常發(fā)生腎鈣沉著癥,很可能促使晚期發(fā)生腎功能不全,極少部分可發(fā)生終末期腎病[12]。III型是Bartter綜合征的經(jīng)典類型,通常在學(xué)齡期甚至成年后起病。該類型可能系髓袢升支粗段細胞中存在冗余的氯通道,其臨床癥狀通常不太嚴重。后期出現(xiàn)低鉀血癥、代謝性堿中毒和高鈣尿癥。IV和IVb型Bartter綜合征存在聯(lián)合缺陷,會同時累及CIC-Ka和CIC-Kb兩種通道,導(dǎo)致嚴重疾病,通常有產(chǎn)前表現(xiàn)和先天性聽力損失。兩種氯通道對內(nèi)耳血管紋的正常離子傳輸和建立正常的耳蝸內(nèi)電位至關(guān)重要[13-15],在耳中該兩種通道功能有重疊,需CIC-Ka和CIC-Kb均缺陷才會出現(xiàn)聽力損失。IV或IVb型Bartter綜合征患者較少發(fā)生腎鈣沉著癥,容易發(fā)生進行性腎功能不全。V型Bartter綜合征亦被稱為常染色體顯性低鈣血癥或常染色體顯性甲狀旁腺功能減退癥,該型系位于染色體3q13 上的CASR 基因突變所致[16-17]。Gitelman綜合征為常染色體隱性遺傳,是位于染色體長臂 16q13 的 SLC12A3 致病基因突變所致其編碼遠端腎小管(DCT)上的噻嗪類敏感性 Na+-CL-共轉(zhuǎn)運體(NCCT)功能失活所致[18-19]。已報道SLC12A3基因有大于430個位點的突變與GS發(fā)病有關(guān),60%為錯義突變,中國人突變類型以T60M為主[20-21]。上述疾病分型有助于臨床醫(yī)師理解其發(fā)病機制,便于基因診斷及治療。
GS與BS系基因突變導(dǎo)致,目前尚無根治的方法,臨床上以控制癥狀為主,給予補鉀、補鎂及聯(lián)合其他藥物應(yīng)用。1、補充電解質(zhì)。通過口服氯化鉀及靜脈補充10%枸櫞酸鉀(2-6 g/d),改善低血鉀及代謝性酸中毒,補鉀目標為3.0 mmol/L以上[22];對于GS低鎂患兒鼓勵進食含鎂豐富的食物如堅果、黑巧克力等。藥物補鎂首選口服,當患者存在嚴重并發(fā)癥或不能耐受口服補鎂時,需靜脈補鎂治療,常用有氯化鎂、門冬氨酸鉀鎂等推薦劑量3mmol/m2/d或 4-5 mg/kg/d,補鎂目標為0.6 mmol/L以上。2、潴鉀類利尿劑(醛固酮拮抗劑):螺內(nèi)酯(推薦劑量為 60 -180 mg/d)可拮抗醛固酮活性,減少尿鉀排泄從而升高血鉀,但在應(yīng)用過程中需注意其抗雄激素的副反應(yīng),如月經(jīng)紊亂、多毛癥、男性乳腺發(fā)育等,同時注意補充鈉鹽以防發(fā)生低血壓,選擇性醛固酮拮抗劑依普利酮副反應(yīng)相對較少[23]。3、腎素一血管緊張素拮抗劑[血管緊張素轉(zhuǎn)化酶抑制劑(ACEI)/血管緊張素Ⅱ受體拮抗劑(ARB)]:可抑制RAAS活性,首選ACEI類藥物,從小劑量遞增,在腹瀉、嘔吐等急性失鈉的情況下不宜選用。4、前列腺素合成酶抑制劑:前列腺素E2(PGE2)能提高RAAS系統(tǒng)的活性,加速鉀離子的流失,同時使ROMK通道的活性降低,導(dǎo)致NKCC2的功能喪失,致使通過致密斑的離子及其相鄰部位的遠端小管上皮細胞內(nèi) CL-減少,最終使COX表達增多,首選吲哚美辛(推薦劑量為 1.5-2.5 mg/( kg.d ),劑量在3 mg/( kg.d )以下為安全劑量,最大劑量不大于5 mg/( kg . d )[24-25],一般建議4-6周以后的患兒使用,早期使用不但效果欠佳,且會增加發(fā)生壞死性小腸結(jié)腸炎的風(fēng)險[26]。多數(shù)GS患者血中PGE2水平正常,故該藥在GS患者中應(yīng)用較少。選擇性COX2抑制劑可治療頑固性低鉀血癥,但需注意其長期心血管副作用[27]。5、腎移植。腎移植可糾正Gitelman綜合征和Bartter綜合征中的轉(zhuǎn)運異常,目前未見移植后疾病復(fù)發(fā)報道,但只有極少數(shù)患者實施腎移植。6、分子伴侶試驗性治療。部分導(dǎo)致Gitelman綜合征和Bartter綜合征的突變會產(chǎn)生功能正常的轉(zhuǎn)運蛋白,但突變會導(dǎo)致這些轉(zhuǎn)運蛋白滯留在細胞內(nèi),不能正確插入合適的細胞膜,使用分子伴侶(如4-苯丁酸)可以改善這些有全部或部分功能的蛋白質(zhì)向細胞膜運動和插入,從而部分挽救氯化鈉的重吸收[28-30]。7、中藥治療。國內(nèi)有學(xué)者提出使用中藥來治療該類疾病,但其療效需更多臨床進一步研究證實[31-32]。
本研究2例患者均系因其他疾病住院治療期間發(fā)現(xiàn)頑固性低血鉀,通過臨床表現(xiàn)及基因測序結(jié)果明確診斷,在規(guī)范補液及螺內(nèi)酯治療后均能維持電解質(zhì)及酸堿平衡,低鎂血癥、低尿鈣是Gitelman綜合征區(qū)別于Bartter綜合征的重要臨床表現(xiàn)。該類病極容易漏診、誤診,作為基層醫(yī)院醫(yī)師,應(yīng)該提高對GS/BS的認識,在臨床上遇到頑固性低鉀血癥、低鎂血癥、低尿鈣及立、臥位腎素-醛固酮增高的患者需仔細詢問病史及家族史,在排除藥物及其他影響后應(yīng)懷疑本類病,盡早行基因檢測明確診斷,同時對其家系基因進行篩查,提高診斷率。并告知家屬GS/BS 需終生治療,強調(diào)個體化管理,并定期隨訪檢測調(diào)整用藥,減少遠期并發(fā)癥,提高患者的生活質(zhì)量。
參考文獻:
[1] Bartter FC, Pronove P, John R, et al. Hyperplasia of the juxtaglomerular complex with hyperaldosteronism and hypokalemic alkalosis[J]. Journal of the American Society of Nephrology, 1998, 9(3):516-528.
[2] Gitelman, H J , Graham, J B , & Welt?, LG . A familial disorder characterized by hypokalemia and hypomagnesemia*[J]. Annals of the New York Academy of Sciences,1969, 162(2), 856-864.
[3] Melander O , Orho-Melander M , Bengtsson K , et al. Genetic Variants of Thiazide-Sensitive NaCl-Cotransporter in Gitelman\"s Syndrome and Primary Hypertension[J]. Hypertension, 2000, 36(3):389-394.
[4] Naomi T , Yoshihiro K , Nozomu I , et al. A High Prevalence of Gitelman's Syndrome Mutations in Japanese[J]. Hypertension Research, 2004, 27(5):327-331.
[5] Zhong F, Ying H, Jia W,et al. Characteristics and Follow-Up of 13 pedigrees with Gitelman syndrome.J. Endocrinol. Invest.2019 ,42(6):653-665.
[6] Miya, A., et al., Gitelman's syndrome with hyperphosphatemia, effectively responding to single oral magnesium oxide administration: A case report. Medicine (Baltimore), 2019. 98(28): p. e16408.
[7] 馬駿,任紅,謝靜遠,等.Gitelman綜合征47例臨床特征分析 [J].中國實用內(nèi)科雜志,2014,34(3):273-276.
[8] Ji W , Foo J N , O"Roak B J , et al. Rare independent mutations in renal salt handling genes contribute to blood pressure variation[J]. NATURE GENETICS, 2008, 40(5):592-599.
[9] 沈曉明. 臨床兒科學(xué)[M] . 2 版. 北京: 人民衛(wèi)生出版社,2013:712-714
[10] Blanchard A , Bockenhauer D , Bolignano D , et al. Gitelman syndrome: consensus and guidance from a Kidney Disease: Improving Global Outcomes (KDIGO) Controversies Conference[J]. Kidney International, 2017, 91(1):24-33.
[11] Finer G , Shalev H , Birk O S , et al. Transient neonatal hyperkalemia in the antenatal (ROMK defective) Bartter syndrome[J]. Journal of Pediatrics, 2003, 142(3):0-323.
[12] Brochard K , Boyer O , Blanchard A , et al. Phenotype-genotype correlation in antenatal and neonatal variants of Bartter syndrome[J]. Nephrology Dialysis Transplantation, 2009, 24(5):1455-1464.
[13] Kontorinis G , Giesemann A M , Iliodromiti Z , et al. Treating hearing loss in patients with infantile Bartter syndrome[J]. The Laryngoscope, 2012, 122(11):2524-2528.
[14] Estévez, Raúl, Boettger T , Stein V , et al. Barttin is a Cl- channel β-subunit crucial for renal Cl- reabsorption and inner ear K+ secretion[J]. Nature, 2001, 414(6863):558-561.
[15] Janssen A G H , Scholl U , Domeyer C , et al. Disease-Causing Dysfunctions of Barttin in Bartter Syndrome Type IV[J]. Journal of the American Society of Nephrology, 2009, 20(1):145-153.
[16] Jentsch T J , Maritzen T , Zdebik A A . Chloride channel diseases resulting from impaired transepithelial transport or vesicular function[J]. Journal of Clinical Investigation, 2005, 115(8):2039-2046.
[17] Lang F , Vallon V , Knipper M , et al. Functional significance of channels and transporters expressed in the inner ear and kidney[J]. AJP: Cell Physiology, 2007, 293(4):C1187-C1208.
[18] Shibli A A , Narchi H . Bartter and Gitelman syndromes: Spectrum of clinical manifestations caused by different mutations[J]. World Journal of Methodology, 2015, 5(2):55-61.
[19] 周建華. 失鹽性腎小管病診斷和治療的研究進展[J]. 臨床腎臟病雜志, 2018, 18(11):6-9.
[20] Wang, Fen, Shi, Chuan, Cui, Yunying,等. Mutation profile and treatment of Gitelman syndrome in Chinese patients[J]. Clinical & Experimental Nephrology, 2017, 21(2):293-299.
[21] 邵樂平, 逯靜茹, 郎艷華, et al. Genotype, phenotype, and follow-up of Chinese patients with Gitelman's syndrome%中國 Gitelman 綜合征患者的基因型、表型分析及隨訪研究[J]. 中華內(nèi)分泌代謝雜志, 2017, 033(001):40-46.
[22] Gitelman綜合征診治專家共識協(xié)作組. Gitelman綜合征診治專家共識[J]. 中華內(nèi)科雜志, 2017, 56(9):712-716.
[23] Morton A . Eplerenone in the treatment of Gitelman's syndrome[J]. Internal Medicine Journal, 2008, 38(5):377.
[24] Georges Deschênes, Fila M . Primary Molecular Disorders and Secondary Biological Adaptations in Bartter Syndrome[J]. International Journal of Nephrology, 2011, 2011(3):1-7
[25] Knoers N V A M . Inherited forms of renal hypomagnesemia: an update[J]. Pediatric Nephrology, 2009, 24(4):697-705.
[26] 王成月,劉利,李小雙,等. 兒童巴特綜合征12例臨床分析[J]. 安徽醫(yī)藥, 2018.22(1):71-73.
[27] Mayan H , Gurevitz O ?, Farfel Z ?. Successful treatment by cyclooxyenase-2 inhibitor of refractory hypokalemia in a patient with Gitelman's syndrome[J]. Clinical Nephrology, 2002, 58(1):73-76.
[28] Andrini O , Keck M , Briones R , et al. ClC-K chloride channels: emerging pathophysiology of Bartter syndrome type 3[J]. American Journal of Physiology - Renal Physiology, 2015, 308(12):F1324-F1334.
[29] De Jong J C , Willems P H G M , Goossens M , et al. Effects of chemical chaperones on partially retarded NaCl cotransporter mutants associated with Gitelman\"s syndrome in a mouse cortical collecting duct cell line[J]. Nephrology Dialysis Transplantation, 2004, 19(5):1069-1076.
[30] Peters M , Ermert S , Jeck N , et al. Classification and rescue of ROMK mutations underlying hyperprostaglandin E syndrome/antenatal Bartter syndrome[J]. Kidney International, 2003, 64(3):923-932.
[31] Luo JW, Meng XR, Yang X, et al. Analysis of mutations of two Gitelman syndrome family SLC12A3 genes and proposed treatments using Chinese medicine[J]. Chin J Integr Med, 2017(6):461-468.
[32] Luya R , Zhijuan D , Yingying Z , et al. Clinical features and gene mutation analysis of two young patients with Gitelman syndrome[J]. Zhejiang Mdical Journal, 2018.40(1):23-26.
作者簡介:劉敏(1991-09),女,碩士,住院醫(yī)師,從事兒科臨床研究,1031768495@qq.com.]
