基于三種貝葉斯方法的結核病基因數據挖掘及生物信息學分析
2021-11-26 06:54:42吳佩望
工程數學學報 2021年5期
張 旭, 吳佩望, 喬 峰
(西南大學數學與統計學院,重慶 400715)
1 引言
到目前為止,結核病仍然是全世界人類發病和因病死亡的主要原因,全世界每年有超過1000 萬人感染結核病,160 余萬人因結核病而死亡,更是有四分之一的人口是潛伏性結核病患者.目前中國乃至全世界在結核病防治問題上依然存在著很大的困難,如結核病發現率低、病原篩查率低、確診率低、致病細菌耐藥率高等.
目前我國對結核病的診斷主要通過痰涂片和病原分離培養.而痰涂片采用的方法靈敏度和特異度都較低,且受實驗室條件及實驗人員的影響較大.病原體分離培養則耗時較長,快速生長菌也至少需要3 天才有結果,時間長者甚至達到數周[1].因此,利用數據分析方法探索結核病發病機制,為疾病防控提供理論依據,并促進了結核病感染的分子診斷及治療技術的發展.
人類全血以及外周血單核細胞的基因組表達分析已被廣泛應用于檢測活動性肺結核患者的宿主轉錄反應,并識別用于診斷的潛在生物標記物.例如:針對本文所用到的數據集,Cai 等[2]應用全基因組轉錄微陣列分析方法測定了結核病患者和對照組外周血單核細胞基因的表達量,并通過qRT-PCR 方法驗證C1q 表達,發現與健康對照組和潛伏性結核感染患者相比,活動性結核病患者外周血中C1q 表達顯著增加,證明了C1q 表達與人類結核中的活動性疾病相關,可能是區分活動性與潛伏性結核病以及結核性胸膜炎與非結核性胸膜炎的潛在診斷標志物.Blankley 等[3]對活動性結核病與健康對照組之間進行Benjamini Hochberg 多重檢驗校正的獨立t檢驗鑒別了380 個差異表達的基因.Alam 等[4]利用Limma 包在健康組和活動性結核病患者之間鑒定出了266 個差異表達基因,其中149 個上調,117 個下調;在活動性結核病和潛在感染組之間共發現127 個上調基因和69 個下調基因.
然而,生命現象的復雜性與不確定性使得傳統統計方法的使用存在很大的局限性,經典統計學方法一系列嚴格的前提假設并不能被很好的滿足.比如,t檢驗往往需要在數據服從正態分布的假設下使用,但基因數據多數都不滿足該假設,盲目使用將會使得研究結果與實際情況相去甚遠.對于數據分析結果的驗證目前也是一個棘手的問題.前面的這幾篇文獻對于選出的基因有的并沒有做進一步驗證,有的采用了qRT-PCR 方法驗證,而該方法依賴于實驗,耗時長、費用高,一般只能對極少數基因進行驗證.因此,為了更加準確地模擬及預測具體的生物學過程,迫切需要一套基于數學、統計學以及計算機科學等的完整有效的組合方法,從海量的數據資源入手挖掘出隱含的、有價值的信息,與微生物學領域的研究方法形成互補,這也正是本研究的主旨.
本文先通過兩種基于貝葉斯統計框架的方法,即線性模型及經驗貝葉斯方法和信息先驗性貝葉斯檢驗方法相結合,篩選出了結核病的潛在易感基因.再經過樸素貝葉斯分類器驗證了這些易感基因的準確性,突出了貝葉斯方法在基因數據分析中的重要性.然后對這些基因進行了生物信息學分析,從生物學角度分析了結核病發病的分子機制.以期為結核病的診斷、長期防控工作提供參考依據.
2 數據和方法
2.1 數據來源及處理
本文所使用的數據集是來自美國國立生物技術信息中心(National Center for Biotechnology Information, NCBI)的高通量芯片表達譜數據庫中的GSE54992,其中包含了9 例結核病患者樣本(TB),12 例結核病潛伏患者樣本和6 例正常樣本(HD),每個樣本有22283 個探針.該數據集應用全基因組轉錄微陣列分析方法測定了結核病患者和對照組外周血單核細胞基因的表達量[2].
本文還選取了NCBI 的另一組數據,同樣來自NCBI 的高通量芯片表達譜數據庫中的GSE83456 用作差異表達基因的驗證,其中包含了45 例結核病患者樣本(PTB)與61 例正常樣本(HC),每個樣本有47231 個探針.該數據集應用全基因組轉錄微陣列分析方法測定了結核病患者和對照組外周血單核細胞基因的表達量[3].
這兩個數據集均已經過標準化,可以直接用于分析.在將探針轉化為基因符號時,我們將對應同一基因符號的多個探針中的表達量的最大值作為該基因的表達量.
2.2 研究方法
2.2.1 線性模型及經驗貝葉斯方法
線性模型及經驗貝葉斯方法(Limma)是由Smyth 根據文獻[5]提出的層次模型發展而來的,針對基因組微陣列數據“大p小n”問題的解決方法[6].Limma 整合了多種統計原理,它在基因表達值矩陣上運行,矩陣中每一行代表一個基因或者探針,每一列對應一個樣本.一方面,通過加權或廣義最小二乘法擬合多個線性模型,并以各種方式利用這些模型的靈活性.另一方面,由于基因組數據的高度平行性,它在每一個基因模型之間借用信息,允許基因之間及樣本之間存在不同程度的差異,這使得統計結論在樣本量較小的數據集中更為可靠.Limma 證明可以使用經驗Bayes 后驗方差估計進行精確的小樣本推斷.這種方法在小樣本的實驗中被證明是特別有優勢的,確保了即使在重復次數很少的情況下,也能得到可靠和穩定的統計推斷.Limma 是選取差異基因的一種基礎的,傳統的方法,該方法雖然緩解了由于樣本量小而導致的推理結果不佳的問題,但是在估計低方差或高方差的基因時會引入偏差.
2.2.2 信息先驗性貝葉斯檢驗
信息先驗性貝葉斯檢驗(IPBT)是由Li 等[7]于2015 提出的一種替代方法,它在某種意義上與基于貝葉斯層次模型的Limma 方法“垂直”.Limma 方法中使用了經驗貝葉斯方法借用同一次實驗中不同基因的信息,而IPBT 方法則是借用在過去不同實驗中相同的基因(或探針),但是使用相同的技術,相同類型的芯片,在同一類型的細胞上的測量信息.該方法的關鍵思想是基于豐富的歷史數據為每個基因(或探針)指定了特異的信息性先驗分布然后再進行貝葉斯假設檢驗.因為不同的基因具有不同的生物學功能,所以通常情況下它們的表達量會顯示出相當多樣化的分布情況.IPBT 的模型為
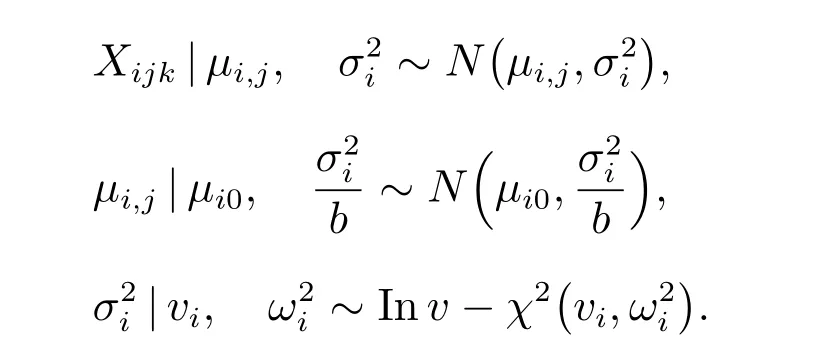

因此,與Limma 在同一實驗中借用不同基因的策略不同,IPBT 假設每個基因都有屬于自己獨特的先驗分布,然后在一次實驗中借用這些歷史數據中的先驗信息來分析.經過與仿真數據的分析對比并繪制ROC 曲線,這種方法被證明篩選結果優于SAM、t檢驗及Limma 等其他方法[8].
2.2.3 樸素貝葉斯分類器
樸素貝葉斯是一種構造分類器的簡單技術,是在貝葉斯算法的基礎上進行相應的簡化,即假設給定目標值時屬性之間相互條件獨立.也就是說沒有哪個屬性變量對于決策結果來說占有著較大或者較小的比重.雖然這個簡化方式在一定程度上對貝葉斯分類算法的分類效果有稍許影響,但是在實際的應用場景中,極大地簡化了貝葉斯方法的復雜性[8].樸素貝葉斯主要基于條件概率模型:給定一個要分類的問題,用向量x= (x1,x2,1,··· ,xn)代表n個特征,對于K個可能的類別中的每一類Ck,使用貝葉斯定理,將每一類的概率寫為

其中Ck類變量的條件分布是
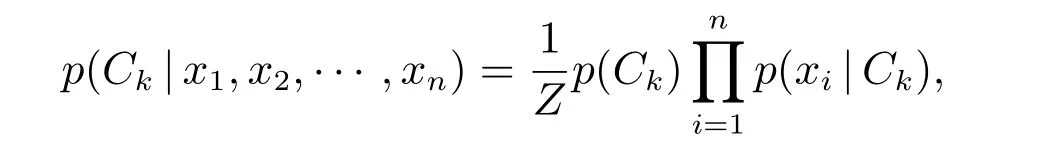
Z是一個只依賴于特征x的縮放因子,當特征變量的值已知時是一個常數.然后根據建立的樸素貝葉斯概率模型和最大后驗概率決策準則構造樸素貝葉斯分類器[9]
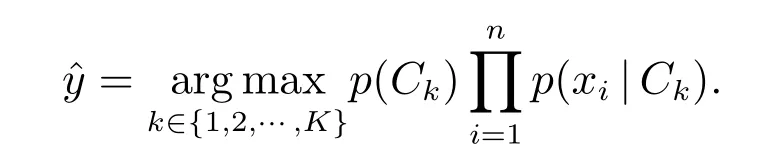
樸素貝葉斯分類器具有穩定的分類效率,在小規模數據集上的表現很好,能夠處理多分類任務,并且對于缺失數據不敏感,在理論上樸素貝葉斯分類器與其他分類方法相比具有最小的誤差率.
2.2.4 生物信息學分析
Gene Ontology(簡稱GO)是一個國際標準化的基因功能分類體系,是一項重要的生物信息學計劃.而GO 富集分析是一種利用基因本體論分類系統解釋基因組的技術,其中基因根據其功能特征被分配給一組預定義的領域,如細胞組分、分子功能、生物過程[10].本文中,我們利用文獻[11]提供的R 語言包clusterProfiler 中的enrichGO 函數進行了GO 功能富集分析,并將其結果可視化.對差異表達基因進行生物信息學分析的另一種方案是KEGG 數據庫[12],其中我們將根據基因所涉及的通路進行分組,并通過假設檢驗分析這些差異表達基因是否在某些通路上存在富集.本文中我們利用文獻[11]提供的R 語言包clusterProfiler 中的enrichKEGG 函數進行了KEGG 通路分析.
3 結果與分析
3.1 篩選結果
GSE54992 數據集經過合并探針后,我們共得到13516 個基因的表達量數據.本文僅選取了其中的結核病患者和正常人兩種樣本數據進行對比分析.經過繪制PCA 圖,如圖1 所示,可以看出兩組樣本被明顯分開,使我們有可能通過數據對比尋找差異基因.
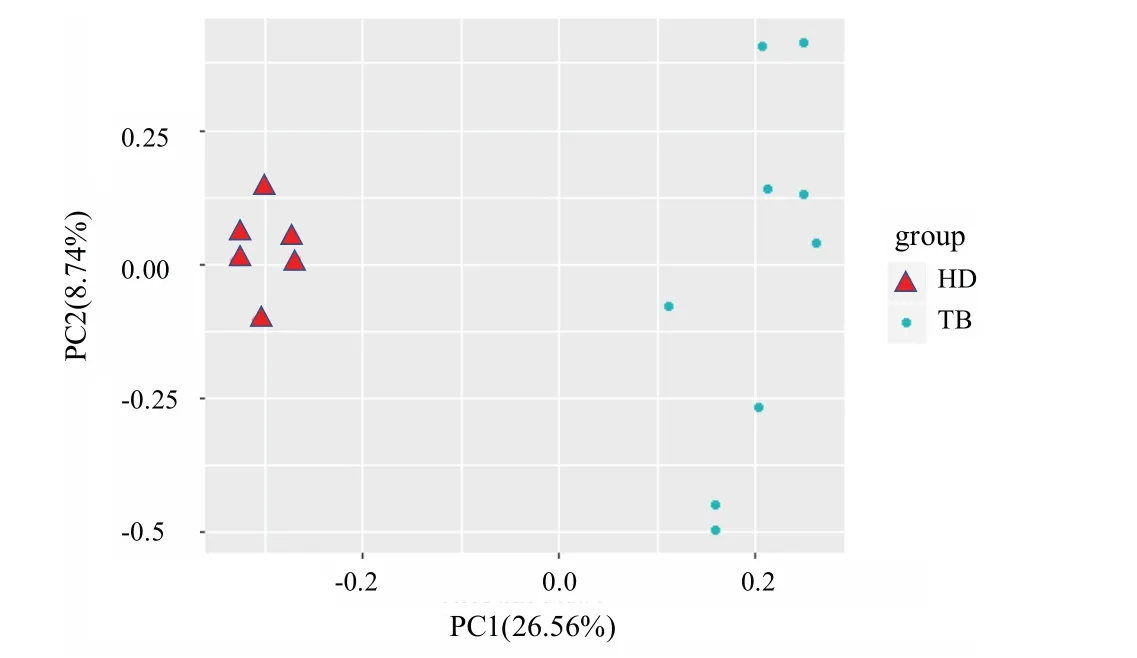
圖1 GES54992 兩組樣本的PCA 圖,HD 為正常樣本,TB 為患病樣本
我們先使用R 語言中的Limma 包處理基因表達量數據,一共得到558 個差異表達基因,其中279 個為上調表達基因,另外279 個為下調表達基因.接著我們又采用了信息先驗性貝葉斯方法再一次篩選差異基因.基于文獻[7]提供的GitHub 中下載得到IPBT 包,我們一共得到821 個差異表達基因,其中上調基因293 個,下調基因528 個.
為了使篩選的差異表達基因結果更為準確,我們對兩種方法篩選出的差異表達基因取交集,得到了319 個基因,包括114 個上調基因和205 個下調基因.為了能更直觀地看出結果,我們進行了可視化,繪制了這319 個基因的韋恩圖和熱圖,如圖2 和圖3 所示.

圖2 IPBT 與Limma 篩選出的差異表達基因數量韋恩圖,其中319 個基因被兩種方法均識別

圖3 319 個基因及15 個樣本的表達熱圖
圖3 中橫軸是根據患病情況分組的15 個樣本,縱軸是選出的319 個基因,每個小方塊為一個基因的表達量.顏色的深淺代表該基因表達量的高低,紅色越深表明基因表達量越高,藍色越深表示基因表達量越低.從圖中我們可以明顯看出樣本被分為兩個組別,而上調與下調基因之間表達量也存在著很大的差異.表明我們通過這兩種方法篩選出的差異表達基因的效果較好.
3.2 模型驗證
由于數據集GSE54992 的樣本本身的區分度較好(圖1),我們選取了另一組數據GSE83456 來驗證差異基因的分類效果.從圖4 可以看出該數據集的兩種樣本有很大部分相互交疊在一起,難以區分開.

圖4 GSE83456 兩組樣本的PCA 圖,其中HC 為正常樣本,PTB 為患病樣本
對于GSE83456 這個獨立驗證集,我們采用兩種方式選取了特征基因建立樸素貝葉斯分類模型進行對比,其一為IPBT 與Limma 兩種方法共同篩選得到的319 個差異表達基因,其二為隨機選取319 個基因.在70%訓練集,30%驗證集的條件下建立樸素貝葉斯分類模型,通過ROC 曲線我們發現,隨機選取特征基因用作建模的準確率僅為55.6%,而通過基于貝葉斯方法篩選出來的差異表達基因用作特征基因對該數據集進行建模得到的分類準確率超過了85%,遠遠高于隨機選取基因用作特征基因的表現.因此,我們認為篩選出的差異表達基因在結核病患病與否的預測上有較為顯著的作用.
3.3 生物信息學分析結果
我們先將差異表達基因分為上調基因與下調基因,再分別進行了GO 功能富集分析,并根據p值大小分別選出上調和下調的各前十名,共20 項功能.經過文獻查閱,我們發現選出的功能類別中有10 項功能與結核病發病相關,其中包括細胞遷移的正調節[13]、白細胞遷移[13]、髓樣白細胞遷移[13]、細胞趨化性[14]、白細胞趨化性[14]、血管系統發育的調節[15]、血管生成[15]、運動的正調節[16]、細胞成分運動的正調節以及細胞運動的正調節[16].表明我們所篩選出來的差異表達基因中大多數基因被富集在與結核病發病相關的基因功能中.隨后我們對選出的319 個差異表達基因進行KEGG 通路分析.由于無論是上調或是下調的基因均可能產生同一生物效應,或在同一通路中出現,因此,我們使用所有的差異表達基因一起進行KEGG 通路富集分析,通過查閱文獻發現,p值列于前十的通路中,細胞因子-受體相互作用[17]、趨化因子信號通路[18]、抗生素的生物合成[19]與結核病發病相關,結核病與百日咳[20]、軍團病[21]、阿米巴病[22]同屬呼吸道疾病并且存在并發癥的情況,更有文章表明用腫瘤壞死因子抑制劑治療類風濕性關節炎可能會導致患活動性結核病的風險顯著增加[23].此外,在所有p值小于0.05 的通路中,我們發現有9 個基因被富集在編號為hsa05152 的名稱為“結核病”的通路中,這9 個基因分別為:IL1A、CAMK2A、IL1B、SPHK1、NFKB1、IL6、CR1、IL10 和JAK2.
4 結論與討論
本文通過Limma 和IPBT 兩種基于貝葉斯統計的方法從數據集GSE54992 中篩選出319 個差異表達基因;再基于獨立驗證集GSE83456 對篩選出的319 個差異表達基因利用樸素貝葉斯分類器進行驗證,使原本難以分開的兩組樣本得到了較準確地分類,證明了篩選的差異表達基因的準確性,同時突出了貝葉斯方法在基因數據分析中的重要性;最后對這些差異表達基因進行GO 功能富集分析和KEGG 通路分析,找到了多個與結核病相關的功能與通路,使篩選出的差異基因得到了進一步驗證.該研究對結核病生物標志物的篩選以及結核病的診斷與防控提供了新的思路.
本文巧妙地組合了三種貝葉斯方法,在傳統的經驗貝葉斯基礎上結合了信息先驗性貝葉斯檢驗來篩選差異基因,使得結果更加可靠;對選出的差異基因又利用樸素貝葉斯分類器進行驗證,通過基因指導結核病樣本分類的高準確率說明了選出基因的重要性.每個步驟都是基于貝葉斯框架,有利于減少系統誤差.此外,IPBT 方法還有很大的提升空間,例如,目前IPBT 提供的歷史數據僅有來自GPL96 平臺的數十種細胞的微陣列數據,今后可以更多地收集相關歷史數據以獲得更準確的差異表達分析結果.
猜你喜歡
音樂探索(2022年2期)2022-05-30 21:01:37
民用飛機設計與研究(2020年4期)2021-01-21 09:15:02
小天使·一年級語數英綜合(2019年8期)2019-08-27 02:23:00
電子制作(2018年18期)2018-11-14 01:48:24
小學科學(學生版)(2018年7期)2018-08-13 09:33:04
山東工業技術(2016年15期)2016-12-01 05:31:22
Coco薇(2016年2期)2016-03-22 02:42:52
Coco薇(2015年1期)2015-08-13 02:47:34
小雪花·成長指南(2015年4期)2015-05-19 14:47:56
鄭州大學學報(醫學版)(2015年2期)2015-02-27 14:50:46
