共濟失調毛細血管擴張癥1例報告并文獻復習
2021-12-17 03:11:08王麗,田剛
中風與神經疾病雜志 2021年10期
關鍵詞:癥狀
王 麗, 田 剛
共濟失調毛細血管擴張癥(ataxia telangiectasia,AT)是一種較少見的常染色體隱性遺傳病,發病率為(0.5~1.0)/10萬人口。它是累及神經、血管、皮膚、單核巨噬細胞系統、內分泌系統的原發性免疫缺陷病,主要表現為小腦性共濟失調,及球結膜、面頰、外耳、頸部等部位的毛細血管擴張,以及反復的呼吸道感染伴發腫瘤傾向。筆者現將孝感市中心醫院神經內科收治的1例AT患者的診治經過報道如下,并復習相關文獻,以期提高臨床同道對AT的認識。
1 臨床資料
患者,男性,15歲,因“行走不穩7 y余”于2019年10月5日入院。患者于7 y前起突然出現頭后仰及背伸,行走不穩,呈間斷性發作,每次持續約1~2 s緩解,發作時無意識障礙及面色青紫,無肢體抽搐及大小便失禁。發病1 y后患者出現持續性的頭后仰、全身左右扭動,不能正常行走,遂到當地醫院完善頭部CT及腦電圖檢查,均未見明顯異常。而后患者全身不自主運動癥狀逐漸加重,曾口服美多巴及安坦治療,癥狀無明顯緩解,且聲光刺激及情緒改變時上述癥狀可加重。既往史:患者足月順產,按計劃預防接種,否認藥物過敏史及輸血史。患者從半歲至9歲期間頻繁發熱,每半月至1 m內即有一次高熱,無高熱驚厥。1歲3個月時有雙足內翻,行走不穩。3歲時患麻疹,已治愈。3歲時發現肚臍左下方皮下腫物,手術切除后送武漢兒童醫院病檢為T細胞淋巴瘤,術后化療一次。體格檢查:神志清楚,全身皮膚、鞏膜無黃染,淺表淋巴結未及腫大。雙側瞳孔等大等圓,直徑3.0 mm,對光反射靈敏,眼球活動自如,可見水平及垂直性眼震,眼結膜毛細血管擴張充血(見圖1)。雙側鼻唇溝對稱,伸舌居中,軟腭居中,咽反射存在。頸抵抗3橫指,左下肢5-級,余肢體肌力5級,四肢肌張力低,肱二頭肌反射存在,余深反射未引出,雙側病理征陽性,指鼻試驗及跟膝脛試驗(+),感覺正常。雙足下垂,內翻畸形(見圖2)。胸前有4.5 cm×3.0 cm的白斑(色素脫失斑)(見圖3)。心肺腹未見明顯異常。輔助檢查:血常規示白細胞計數3.87×109/L,紅細胞計數5.41×1012/L,血紅蛋白119 g/L,平均紅細胞體積69.0 fl,紅細胞平均血紅蛋白濃度318 g/L,血小板計數340×109/L,淋巴細胞計數0.78×109/L;血尿代謝篩查未見明顯異常;血銅藍蛋白正常;血甲胎蛋白200.75 ng/ml(明顯升高);頭部MRI示小腦、兩側橋臂萎縮改變(見圖4);腦電圖正常,聽覺、視覺誘發電位正常;韋氏智力量表79分,社會生活能力評定66分;流式細胞分析總T淋巴細胞、總B淋巴細胞偏低,IgA、IgM正常,IgG偏低;腦脊液常規、生化、免疫相關檢查正常;線粒體基因3243位點、8344位點、8993位點未見異常;肌電圖示神經源性損害。診斷:AT。告知患者家屬病情后,家屬簽字自動辦理出院;后電話隨訪,患者因反復的呼吸系統感染于呼吸科抗感染治療。
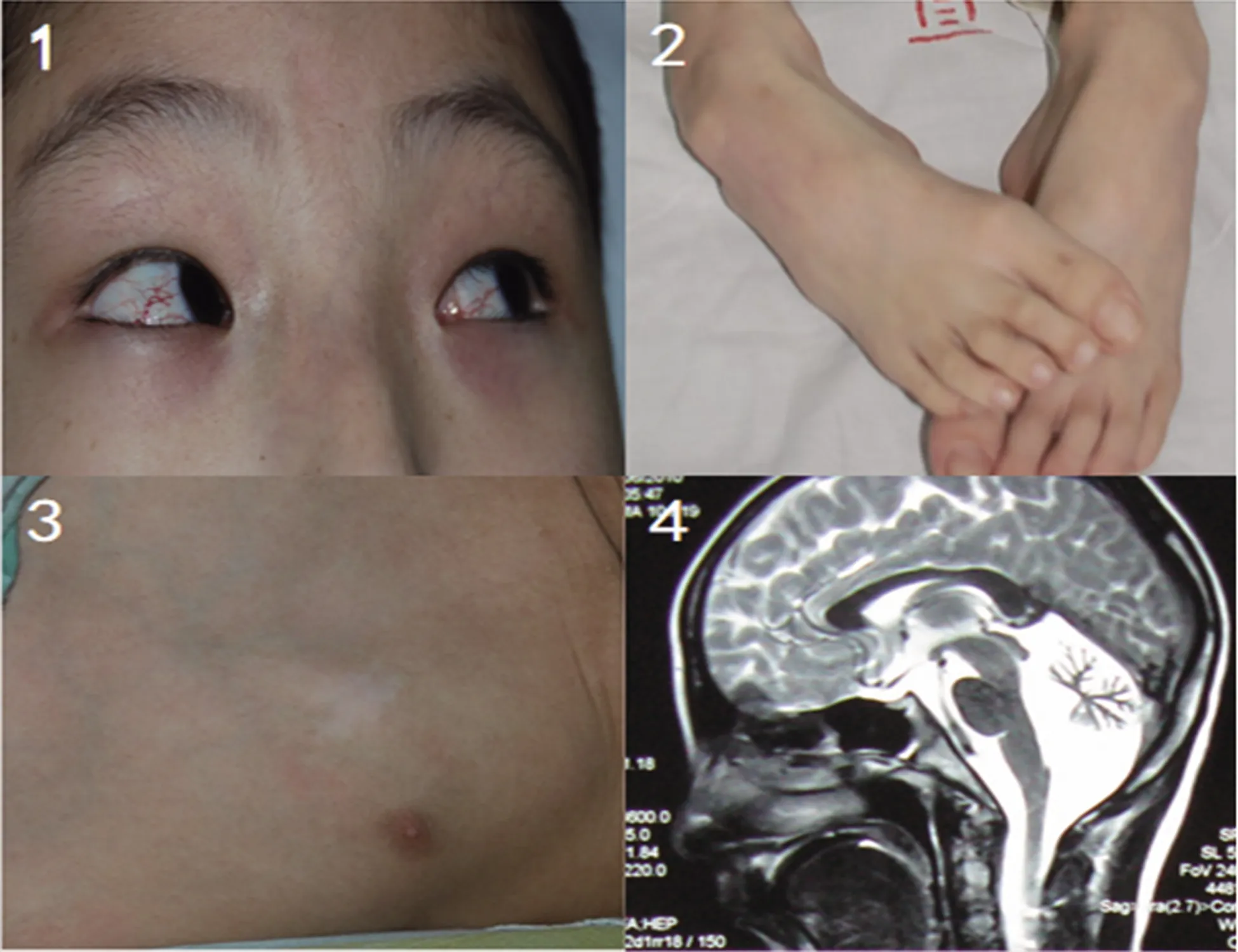
圖1 球結膜毛細血管擴張;圖2 雙足內翻畸形;圖3 胸前色素脫失斑;圖4 頭部MRI示小腦、兩側橋臂明顯萎縮
2 討 論
AT的臨床特征包括:(1)神經系統表現:最初表現為兒童期共濟失調,起病最初表現為學步時笨拙和不穩[1],在小學階段,行走變得更加困難,需要墻壁來支撐,隨后可能需要輪椅。由于眼睛運動協調受損,使得閱讀變得非常困難,并隨后出現構音障礙[1]。同時,在AT患者中,除小腦共濟失調外,舞蹈病和肌張力障礙也是最常見的運動障礙。肌陣攣、震顫和帕金森病的發生率較低[2]。遠端至近端肌腱反射喪失也是AT的特征,反映了進行性的感覺和運動神經病變[3]。(2)神經影像學表現:MRI是AT患者的首選檢查方法,可發現進行性和彌漫性小腦萎縮,但研究發現,萎縮的嚴重程度與臨床特征無明顯的相關性。除了小腦萎縮外,MRI還發現了患者的腦白質異常,包括含鐵血黃素沉積和腦深部毛細血管的擴張[4]。(3)毛細血管擴張:眼球結膜內的毛細血管擴張通常發生在5~8歲,易被誤診慢性結膜炎或過敏。毛細血管擴張也可以出現在暴露在陽光下的皮膚區域,特別是雙耳部、頸部暴露部位、鼻翼和頰部,呈蝴蝶形。(4)視力和眼球運動:毛細血管擴張不影響視力,然而對眼睛運動和視覺固定的控制往往會受到損害,包括:動眼功能減退,眼球震顫和前庭-眼球反射(VOR)異常。(5)免疫系統表現:大約2/3的AT患者有免疫系統的異常[5],最常見的是一個或多個免疫球蛋白(IgG、IgA、IgM或IgG亞類)的水平降低,不能對疫苗或感染產生抗體,淋巴細胞減少,特別是T淋巴細胞。一小部分患者也可能有IgM水平升高,并伴有IgG和(或)IgA缺乏,這可與幼兒期的高IgM綜合征相混淆[6]。AT患者罹患自身免疫性或慢性炎癥性疾病的風險增加,如免疫性血小板減少癥(ITP)、關節炎、白癜風和慢性皮膚肉芽腫等。(6)呼吸系統表現:有25%以上的AT患者會出現慢性肺部疾病[7],患者可表現為持續咳嗽、肺部充血和喘息癥狀,這些癥狀可發生在沒有其他全身癥狀的情況下,導致診斷和治療延遲,最終導致支氣管擴張、復發性肺炎、肺纖維化和間質性肺病(ILD)。其中間質性肺病在AT中最為常見,其特征是肺活量(FVC)減低,而肺活量降低可以增加他們手術麻醉過程中引起的肺部并發癥的風險,因此識別那些患有限制性肺病的患者可以幫助患者減少圍手術期和手術期間的并發癥[8]。(7)癌癥:AT患者癌癥的發病率大大增加[9],淋巴瘤和白血病在年青人中最為常見,隨著年齡的增長,成人也容易罹患其他系統腫瘤,如乳腺癌、肝癌、胃和食管癌等。目前還沒有辦法預測哪一個患有AT的個體會發展為癌癥,如患者出現持續腫大的淋巴結,不明原因的發熱時,必須將血液系統腫瘤視為診斷的可能性。一項2016年的Meta分析發現,在50歲時,攜帶AT基因突變的女性患乳腺癌的累積風險約為6%,在80歲時可上升至30%[10]。(8)輻射的敏感性:AT患者對電離輻射(如X射線和伽馬射線)的敏感性增加,對AT患者是有害的,如必須進行,也應減少劑量[11]。但紫外線照射并不會增加AT患者皮膚癌的發病率,因此不需要對陽光照射采取特殊預防措施。(9)吞咽困難:吞咽困難在AT患者中很常見,因為AT患者的口咽部肌肉運動的協調性下降從而導致咀嚼困難增加吸入性肺炎的風險。(10)內分泌異常:生長發育不良是所有AT患者的一個共同特征,營養不良、感染及生長激素分泌不足是導致生長發育不良的原因[12]。AT患者的胰島素敏感性降低,導致糖尿病[13]。(11)其他的臨床特征:患有AT的人也會有更多的皮膚問題(如白癜風、疣、色素脫失斑等);足部畸形在AT患者中很常見,并且由于協調受損而使得患者行走更加困難。肝臟超聲異常,肝酶升高,脂質代謝改變和CRP升高提示AT有動脈粥樣硬化風險。除了非酒精性脂肪肝會發展為肝硬化和肝癌外,肝臟疾病也可加劇神經系統癥狀,因此,預防肝病對于改善患者的健康,改善其生活質量和耐力特別重要[14]。
ATM基因的突變可引起細胞周期調控及細胞凋亡的異常,從而導致患者出現免疫缺陷、對輻射高敏感性和腫瘤易感性的增加[15]。受到輻射后可發生DNA錯誤修復,這種免疫缺陷狀態可能解釋患者對反復肺部感染及支氣管擴張極顯著的敏感性。因此對于任何患有癌癥的人,如果同時有步態障礙或眼睛有關的不確定障礙運動異常,特別是如果癥狀是進行性的,都應該考慮對AT的診斷。
本病無特效治療,可用大劑量免疫球蛋白靜脈滴注、胸腺肽肌肉注射等可提高患者的免疫功能。目前能采取的唯一有效治療是控制感染。應避免接觸各種射線、烷化物等,防止DNA鏈斷裂。神經問題目前還沒有一種治療方法可以減緩或阻止與AT相關的神經功能缺陷的進展。身體、職業和語言治療以及運動可能會有所幫助,某些抗帕金森病和抗癲癇藥物可能有助于治療癥狀。
猜你喜歡
初中生學習指導·提升版(2023年8期)2023-09-12 10:26:19
保健醫苑(2022年1期)2022-08-30 08:39:40
中老年保健(2021年12期)2021-08-24 03:30:44
今日農業(2020年17期)2020-10-27 03:10:52
今日農業(2020年16期)2020-09-25 03:05:08
家庭醫學(下半月)(2020年2期)2020-05-11 02:07:18
基層中醫藥(2020年10期)2020-02-13 15:45:52
吉林蔬菜(2017年10期)2017-11-01 07:47:04
獸醫導刊(2016年6期)2016-05-17 03:50:35
中國醫學影像學雜志(2015年9期)2015-12-15 11:03:26
