神經元核內包涵體病5例并文獻復習
2021-12-23 06:41:08朱海青周金寶張玉梅嚴春燕
臨床與實驗病理學雜志 2021年11期
宋 坤,柴 學,王 娟,朱海青,周金寶,張玉梅,嚴春燕
神經元核內包涵體病(neuronal intranuclear inclusion disease, NIID)又被稱為神經元核內透明包涵體病(neuronal intranuclear hyaline inclusion disease, NIHID),或者是核內包涵體病(intranuclear inclusion body disease, INIBD)。NIID屬于進展緩慢的神經退行性疾病,其特征是中樞、外周和自主神經系統細胞及體細胞中含有嗜酸性透明核內包涵體。因其臨床表現異質性,易與其他神經系統疾病混淆。本文回顧性分析5例NIID的臨床病理學特征、影像學表現、免疫表型、診斷及鑒別診斷等,旨在提高臨床和病理醫師的認識水平。
1 材料與方法
1.1 臨床資料收集2019年12月~2021年5月南京醫科大學附屬腦科醫院存檔的5例NIID標本,患者男性3例,女性2例;年齡59~70歲,平均66歲。患者均為50歲以后發病;慢性起病4例,亞急性起病1例。
1.2 方法取患者右側外踝上10 cm處皮膚活檢,標本均經10%中性福爾馬林固定,常規脫水、透明、石蠟包埋,4 μm厚連續切片,行HE染色。免疫組化染色及電鏡檢查均由解放軍東部戰區總醫院病理科協助完成。免疫組化采用EnVision兩步法染色,DAB顯色。抗體p62(克隆號SC-28359,稀釋度1 ∶200)購自Santa Cruz公司。免疫組化標記由全自動免疫組化染色儀完成,以胞核呈棕黃色且無背景染色為陽性。電鏡檢查采用2.5%戊二醛和1%鋨酸雙重固定,2.5%戊二醛固定,脫水、環氧樹脂包埋,制成半薄切片,甲苯胺藍染色定位,制成80 nm厚的超薄切片,鉛-鈾雙染色,于透射電鏡(JEM-1011)下觀察。例1和例4進行FMR1基因分析(購自上海昂普生物公司和上海韋翰斯生物公司)。將正常等位基因定義為5~44個CGG重復。
2 結果
2.1 臨床特點5例患者臨床表現復雜多樣,其中記憶力下降為主2例,行走不穩為主2例,意識障礙為主1例。2例患者均以近期記憶力下降為主,查體均顯示記憶力及計算力下降,理解力差,反應遲鈍,1例伴右上肢姿勢性震顫;2例患者中1例認知功能初篩示簡易精神狀態檢查(mini-mental state examination, MMSE)量表為27分、蒙特利爾認知評估(montreal cognitive assessment, MoCA)量表為18分、日常生活活動能力(activity of daily living, ADL)量表為23分,顯示有認知功能障礙;另1例認知功能初篩示MMSE量表為29分、MoCA量表為27分、ADL量表為20分,顯示認知功能尚正常。行走不穩2例,其中1例伴大小便困難及嘔吐癥狀,并伴性格改變;另1例伴肢體抖動、便秘、性格改變等癥狀;伴消化及泌尿系統癥狀的患者呈惡病質狀態,查體示雙下肢肌力4°,伴肢體抖動的患者查體示右側肢體靜止性震顫,上肢明顯,肌張力鉛管樣增高,共濟運動差,右側明顯。意識障礙1例,查體發現其計算力下降,遠近記憶力尚可。本組5例患者有4例無父母系三代家族史,另1例患者母親有“老年癡呆癥”。MRI檢查:DWI示4例雙側大腦半球、1例雙側額頂葉見對稱性皮髓質交界區異常高信號影;T2、FLAIR序列出現腦白質高信號;其中4例腦室有擴大(圖1)。
2.2 病理學檢查5例皮膚活檢標本中,均在真皮層及皮下組織的汗腺細胞、纖維母細胞中見核內嗜酸性透明包涵體。4例在皮下組織的脂肪細胞中見核內嗜酸性透明包涵體,另1例因脂肪細胞極少,未在其脂肪細胞中找到核內嗜酸性透明包涵體(圖2)。
2.3 免疫表型5例NIID標本包涵體中p62均呈陽性(圖3)。
2.4 電鏡檢查5例患者均行電子顯微鏡檢測,在3例患者的皮膚纖維母細胞中見核內包涵體,為類圓形絲狀無膜結構,直徑1.5~3 μm(圖4)。
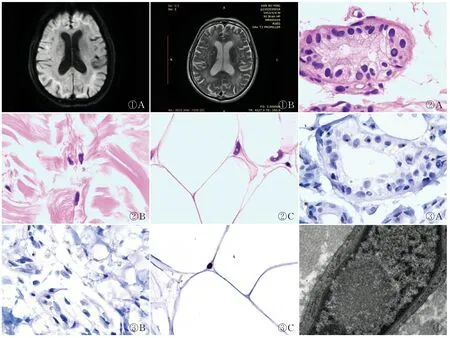
①A①B②A②B②C③A③B③C④
2.5 基因分析2例患者行FMR1基因檢測,CGG重復次數分別為29次和30次,均在正常范圍內(圖5)。
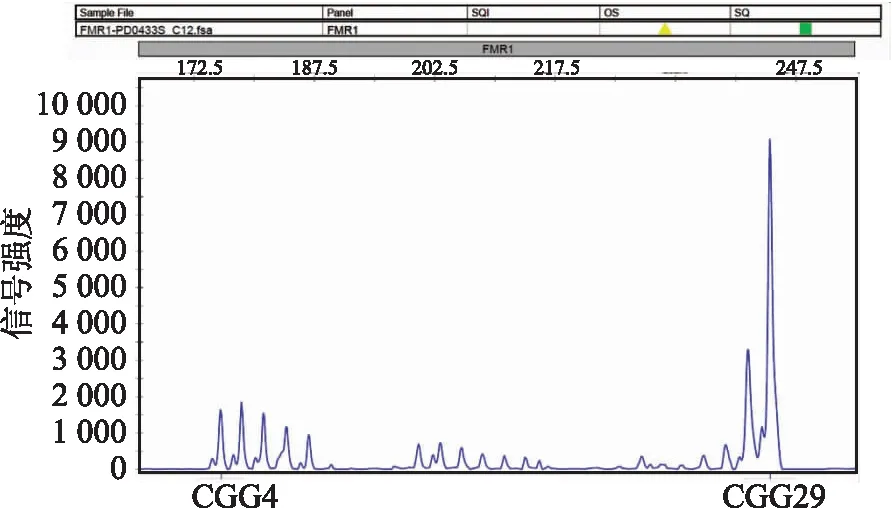
圖5 FMR1基因為CGG重復29次
3 討論
NIID是一種罕見的進展緩慢的神經退行性疾病,其特征是在中樞、外周和自主神經系統的神經元和膠質細胞以及體細胞中含有嗜酸性透明核內包涵體。于1968年由Lindenberg等[1]首次報道,2011年前由于檢查手段的限制,文獻報道較少,隨著皮膚活檢、基因檢測等檢查手段的發展,近年NIID病例報道逐年增多,僅2020年國內外報道合計100余例。
NIID發病年齡跨度較大,從嬰兒到老人(2~78歲)均可發病[2-3]。Takahashi-Fujigasaki等[2]將NIID根據患者發病年齡分為三型:(1)嬰兒型,在嬰兒期發病,臨床病程相對較短(<5歲發病,病程不超過10年);(2)青少年型,在嬰兒期或青少年期發病,臨床病程長達10年或10年以上;(3)成人型,>50歲以后發病。
成人型NIID臨床表現多樣,無特異性,呈慢性或亞急性過程,表現為中樞神經系統以及自主神經受累等。Sone等[4]分析57例成人NIID的臨床和病理特點,根據遺傳學特點將其分為散發型和家族型。散發性NIID患者發病年齡51~76歲,以癡呆癥狀最為突出(94.7%),其次為瞳孔縮小(94.4%)、共濟失調(52.8%)和意識障礙(39.5%)。家族性NIID患者中,發病年齡小于40歲者,以四肢無力最常見(100%),被稱為肢體無力組;在發病年齡超過40歲,以癡呆癥狀最為突出(100%),被稱為癡呆組。Tian等[5]根據臨床表現將家族型NIID又增加新分組 —— 帕金森組。Chen等[6]分析51例NIID患者,發現除神經系統癥狀(行為異常、認知功能障礙、帕金森病、周圍神經癥狀、腦病等)外,其他系統的臨床表現非常顯著且多樣,呼吸系統(刺激性干咳)、消化系統(便秘、惡心嘔吐)、泌尿系統(尿頻、尿急)、循環系統(動脈瘤樣胸痛、體位性低血壓)、生殖系統(前列腺增生癥狀、陰道不規則出血、不孕)、運動系統(肌無力、關節疼痛)等均有不同的臨床表現。因此,Chen等[6]認為系統性核內包涵體病(systemic intranuclear inclusion disease, SIID)比NIID更適合此類疾病的描述。本組5例患者發病年齡均為50歲以上,其中4例無父母系三代家族史,屬于成人散發性病例,而另1例表現為記憶力下降,其母親有癡呆病史,疑似為家族型NIID。5例患者臨床表現各不相同,并且同一患者常伴多種神經系統癥狀以及其他系統癥狀。高度異質的臨床表現,給臨床醫師診斷帶來挑戰。隨著NIID病例的積累,對NIID的臨床表現會有更深入的認識。
NIID的尸檢病例中,發現核內包涵體存在于中樞神經系統的大腦皮質、基底節、腦干和脊髓的神經元及星形膠質細胞、少突膠質細胞中,并伴不同程度神經元丟失,但并不是所有尸檢病例都能見到神經元丟失[2,4,7]。在成人患者中,包涵體主要見于星形膠質細胞[3,7]。具有核內包涵體的星形膠質細胞功能障礙,可能導致腦白質髓鞘和軸突的繼發性損傷,引起海綿樣變,這些細胞的功能紊亂可以改變神經元功能,可能是導致癡呆癥的原因[7]。在內臟器官中,臨床病理相關性尚不完善,自主神經功能障礙最可能是由核內包涵體引起的。Sone等[8]報道2例表現為運動感覺和自主神經病變的NIID尸檢病例,發現在交感神經、腸肌叢神經節神經元、背根神經節神經元及脊髓運動神經元中廣泛存在核內包涵體。本組例1及例4患者便秘,推測可能與其腸肌叢神經節神經元中含有核內包涵體有關。多樣的臨床癥狀是否與NIID的核內包涵體直接相關,需積累更多病例進行分析。
成人型NIID患者影像學表現相對具有特征性,DWI序列上可見沿著皮質-髓質交界區分布的特征性異常高信號病變,在病程初期病變主要局限于額葉,隨著病程進展,DWI高信號逐漸向其他區域發展,但僅限于皮髓質交界,并不向深部白質進展;T2加權像及FLAIR像上的雙側對稱彌漫的腦白質病變,且早期更多以額葉受累為主;腦室擴大。但這些影像學改變并非僅見于NIID,其他多種神經系統性疾病包括各種遺傳代謝相關的腦白質病、脆性X染色體綜合征及腦炎等疾病均可出現類似的影像學表現。Yokoi等[9]認為皮質下白質呈多發性病理性海綿狀改變與影像學上皮質下線性DWI高信號病變有關。雖然Sone等[4]報道的NIID中,100%的散發病例有DWI高信號。有文獻報道[10],患者持續4年復發性嘔吐,DWI顯示無明顯異常,直到7年后右額葉皮質延髓交界處出現輕微異常強度病變。Kawarabayashi等[11]報道1例患者發病5年后DWI高信號消失,推測持續的神經元丟失和膠質增生可能是其消失的原因。DWI高信號常被用作NIID的診斷指征,但不出現高信號或者高信號的消失可能導致NIID的誤診。本組5例均可見DWI序列上雙側對稱性皮髓質交界區高信號,T2、FLAIR序列均出現腦白質高信號,顯示腦白質變性,其中4例伴腦室擴大。
目前,NIID的發病機制尚不清楚。Takahashi-Fujigasaki等[12]研究發現NIID中核內包涵體的形成可能與多聚谷氨酰胺疾病具有相似的病理、生理途徑。NIID和多聚谷氨酰胺疾病中的核內包涵體均是泛素化的,并且含有與泛素-蛋白酶體-蛋白質降解系統相關的其他成分。核內包涵體被認為是由過度積聚在細胞核內的降解蛋白質形成的。未知的異常蛋白的聚集或核內蛋白降解系統的功能障礙,可能NIID是疾病進展的原因。雖然NIID中的神經元核內包涵體對識別異常擴增多聚谷氨酰胺的單克隆抗體1C2具有免疫反應性,但頻率較低。在NIID中沒有發現導致多聚谷氨酰胺擴增的原因,也沒有證據表明NIID是一種多聚谷氨酰胺疾病。2019年,關于NIID分子機制的研究取得重大突破。Sone等[13]在某家族性NIID中發現NOTCH2NLC基因5′區的GGC異常重復擴增,隨后在另外8個家族性NIID及40個散發性NIID中發現相似的擴增。NOTCH2NLC基因是位于1q21.1的三個人類特異性NOTCH2相關基因(NOTCH2NLA、NOTCH2NLB和NOTCH2NLC)之一,其在各種膠質細胞中高表達,被認為與人類大腦皮層的進化擴展有關。2019年,Tian等[5]報道來自4個家族性及5個散發病例中NOTCH2NLC基因GGC重復擴增,并發現家族型NIID中肌無力組GGC重復數為118~517次;帕金森組GGC重復數為66~102次;癡呆癥組GGC重復數為91~268次;而散發病例GGC重復數為86~133次。NIID臨床表現的多樣性可能與NOTCH2NLC基因5′區域GGC病理性重復次數相關,一般認為GGC重復擴增次數超過60次具有致病性。
2011年Sone等[14]提出皮膚活檢有助于神經核內包涵體疾病診斷。此前,診斷NIID通常依靠尸檢、腦活檢或直腸活檢及腓腸神經活檢,費用高,且常發生復雜的穿透性損傷。皮膚活檢是NIID的一種有效的、微創的診斷工具,臨床建議取外踝10 cm處、3 mm厚皮膚活檢進行病理檢查,取至真皮層及皮下脂肪組織,以便觀察到汗腺細胞、纖維母細胞及脂肪細胞中的核內包涵體。光鏡下汗腺細胞、纖維母細胞及脂肪細胞中可見核內嗜酸性透明包涵體,呈圓形,直徑為1.5~10 μm,位于核仁附近[4]。免疫組化染色核內包涵體呈泛素及泛素相關蛋白,如NUB1(NEDD8 ultimate buster 1)、小泛素修飾物-1(small ubiquitin modifier-1, SUMO-1)、小泛素修飾物-2(small ubiquitin modifier-2, SUMO-2)和p62均陽性。本組5例均行外踝10 cm處皮膚活檢,鏡下見真皮層及皮下組織的汗腺細胞、纖維母細胞中找到核內嗜酸性透明包涵體,其中4例在皮下組織的脂肪細胞中找到核內嗜酸性透明包涵體,經免疫組化染色核內包涵體p62呈陽性。電鏡觀察纖維母細胞中有核內包涵體,為類圓形絲狀無膜結構,與文獻報道一致[4,15]。
NIID易與脆性X相關震顫/共濟失調綜合征(fragile X-associated tremor/ataxia syndrome, FXTAS)混淆,其臨床癥狀和病理學表現與NIID類似,且影像學上亦可出現DWI上的皮髓交界區高信號[16],FMR1基因CGG擴增可確診FXTAS。Sone等[17]報道FXTAS中的神經元丟失僅限于浦肯野細胞,并且少突膠質細胞中未觀察到核內包涵體,而NIID中的神經元丟失廣泛分布于中樞神經系統和周圍神經系統,少突膠質細胞中存在p62陽性的核內包涵體。目前,尚未有關于FXTAS患者的皮膚組織學檢查的研究報道。本組例1和例4行FMR1基因檢測,結果顯示CGG重復數均在正常范圍內,可排除FXTAS;另外3例雖未行基因檢測,但典型的影像學表現及皮膚活檢可見細胞核內包涵體,支持NIID的診斷。
目前,NIID尚無有效的治療方法[18],但對于周圍神經受累、癡呆、帕金森綜合征等,對癥用藥可以延緩某些癥狀的發展。現階段尚無大宗臨床病例報道,還需積累更多病例進一步分析。
NIID是一種罕見的進展緩慢的神經退行性疾病,臨床具有高度異質性,易誤診。NIID的診斷需結合特征性的影像學改變,皮膚活檢行HE染色及p62、泛素等免疫組化染色,以及電鏡檢查,必要時可行FMR1基因及NOTCH2NLC基因檢測。
(本文病例承蒙解放軍東部戰區總醫院病理科李南云主任會診及程凱老師提供電子顯微鏡檢查,特此致謝!)
