高效液相色譜法測定雞蛋中氟喹諾酮類藥物殘留量前處理方法的優化
2021-12-31 03:44:56達列亞阿合買提張崇威司慧民宋志超
中國獸藥雜志 2021年11期
陳 薔,達列亞·阿合買提,張崇威,司慧民,張 磊,宋志超*
(1.河南省獸藥飼料監察所,鄭州 450008;2.新疆獸藥飼料監察所,烏魯木齊 830063)
氟喹諾酮類藥物(Fluoroquinolones)是一類人工合成的廣譜殺菌性抗菌藥物,因其具有廣譜、高效、低毒以及與其它抗菌藥物無交叉耐藥性等特點,已廣泛應用于獸醫臨床。但人們若長期使用含較低濃度氟喹諾酮類藥物的動物食品后,易誘導耐藥性的產生[1-2]。目前,雞蛋中氟喹諾酮類藥物殘留的檢測方法有液質聯用法[3-5]、液相色譜法[6-8]、酶聯免疫法[9]等,其中一些是現行的檢測標準[5-7]。在日常檢測中,采用國內農業部781號公告-6-2006和NY 5039-2005的方法對雞蛋中4種氟喹諾酮類藥物檢測時發現回收率較低且平行性較差。本實驗結合幾種常見的氟喹諾酮類藥物檢測標準,優化了樣品提取凈化條件,建立了雞蛋中環丙沙星、達氟沙星、恩諾沙星和沙拉沙星4種氟喹諾酮類藥物多殘留的液相色譜-熒光檢測器分析方法。本方法檢測結果靈敏度高、準確度好,適用雞蛋中氟喹諾酮類藥物殘留的分析檢測。
1 材料和方法
1.1 儀器和材料 高效液相色譜儀Waters2695(配熒光檢測器2475),美國Waters公司;分析天平(感量0.00001 g),METTLE TOLEDO XP205;天平(感量0.01 g),ALC-1100;渦旋混合器,IKA MS2;振蕩器,江蘇金壇市正基儀器有限公司HZ-82;離心機, Sigma公司3-30 K;C18固相萃取柱,美國Agilent公司,規格100 mg,3 mL。
1.2 試劑 乙腈、甲醇均為色譜純;氫氧化鈉、磷酸、三乙胺、磷酸二氫鉀、正己烷為分析純; 實驗用水為高純水;鹽酸環丙沙星、甲磺酸達氟沙星、恩諾沙星、沙拉沙星對照品(中國獸醫藥品監察所,純度≥99%)。
1.3 試劑的配制 5.0 mol/L氫氧化鈉溶液, 取氫氧化鈉20 g或飽和溶液28 mL,加水稀釋至100 mL;0.03 mol/L氫氧化鈉溶液,取5.0 mol/L氫氧化鈉溶液0.6 mL,加水稀釋至100 mL;磷酸鹽緩沖液,取磷酸二氫鉀6.8 g,加水使溶解并稀釋至500 mL(pH≈4.4),用5.0 mol/L氫氧化鈉溶液調節pH值至7.0;0.05 mol/L磷酸/三乙胺溶液,取85%磷酸3.4 mL,用水稀釋至1000 mL(pH≈1.7),用三乙胺調節pH值至2.4)。
1.4 試驗方法
1.4.1 色譜條件 色譜柱:XBridgeTMC18(5 μm,4.6×150 mm);柱溫:30 ℃;流動相:0.05 mol/L磷酸溶液/三乙胺-乙腈(87+13);流速:1.0 mL/min;進樣量:20 μL;檢測波長:激發波長280 nm;發射波長450 nm。
1.4.2 標準曲線繪制 分別稱取4種氟喹諾酮類藥物對照品,用0.03 mol/L氫氧化鈉溶液配制成濃度分別為1 mg/mL(環丙沙星、恩諾沙星、沙拉沙星)和0.2 mg/mL(達氟沙星)的標準貯備液; 用流動相稀釋成濃度分別為2、5、10、20、50、100、200 ng/mL(環丙沙星、恩諾沙星、沙拉沙星)和0.4、1、2、4、10、40 ng/mL(達氟沙星)的系列標準工作液。
1.4.3 樣品前處理 雞蛋去殼,混合均勻。-20 ℃以下貯存備用。稱取新鮮或解凍的試料2±0.02 g置50 mL離心管中,加入磷酸鹽緩沖液10 mL,渦旋混合,中速振蕩10 min,10000 r/min離心10 min,轉移上清液于25 mL容量瓶中。用磷酸鹽緩沖液10 mL重復提取一次。合并兩次上清液,定容,混勻,轉移到50 mL離心管中。加入正己烷10 mL,渦旋混合,中速振蕩5 min。10000 r/min離心10 min,吸取下層清液備用。C18固相萃取柱依次用甲醇2 mL,磷酸鹽緩沖液2 mL預洗。取上述備用液10.0 mL以1 mL/min的速度過柱。用水2 mL洗柱,擠干。用洗脫液2.0 mL以1 mL/min的速度將樣品洗脫入10 mL離心管中,擠干,渦旋混合,過0.22 μm微孔濾膜,供高效液相色譜-熒光檢測器測定。
1.4.4 樣品回收率與精密度 取空白樣品,分別添加5、10、20、50 μg/kg(環丙沙星、恩諾沙星、沙拉沙星)和1、2、4、10 μg/kg(達氟沙星)4個濃度,按1.4.3項進行測定。每個濃度分別測定6次,連續測定4 d,計算回收率及批內批間相對標準偏差。
1.4.5 檢測限 取20個空白樣品進行適當濃度的添加,按上述步驟操作,測得噪音信號的平均值,按信噪比≥3為檢測限。
2 結果與分析
2.1 色譜分離與測定 圖1 為混合標準溶液色譜圖,4種氟喹諾酮類藥物的保留時間依次為環丙沙星5.5 min、達氟沙星6.6 min、恩諾沙星7.7 min、沙拉沙星12.1 min。圖2、圖3分別為雞蛋空白樣品、空白添加樣品的色譜圖。
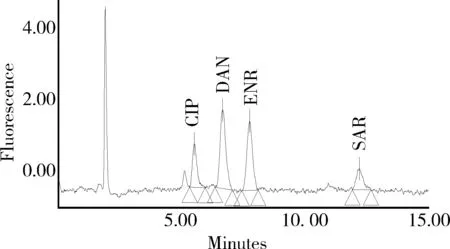
圖1 2 ng/mL標準溶液中氟喹諾酮類藥物色譜圖Fig 1 Chromatogram of standard solution of fluoroquinolones
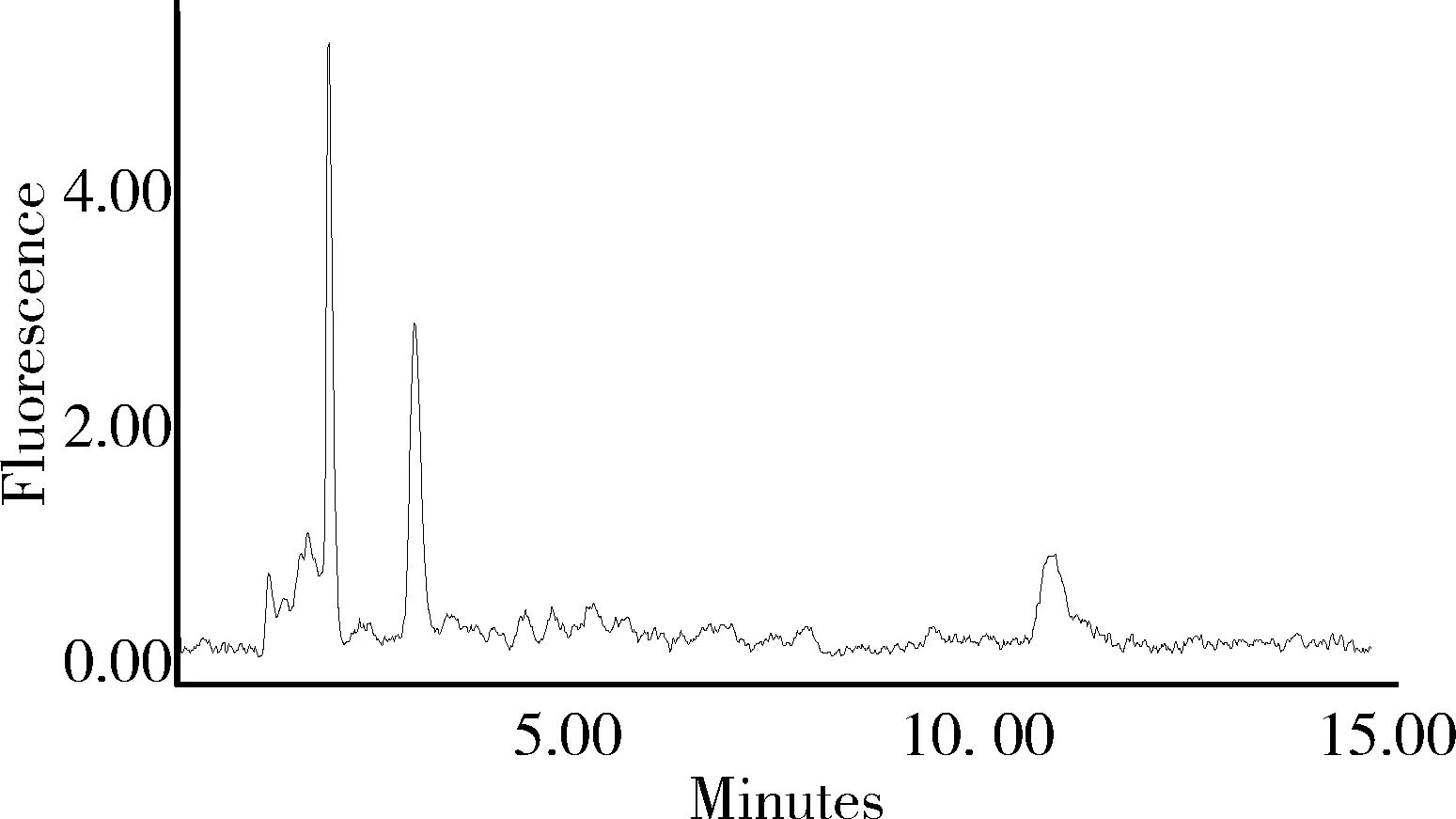
圖2 空白雞蛋中氟喹諾酮類藥物色譜圖Fig 2 Chromatogram of fluoroquinolones in blank egg
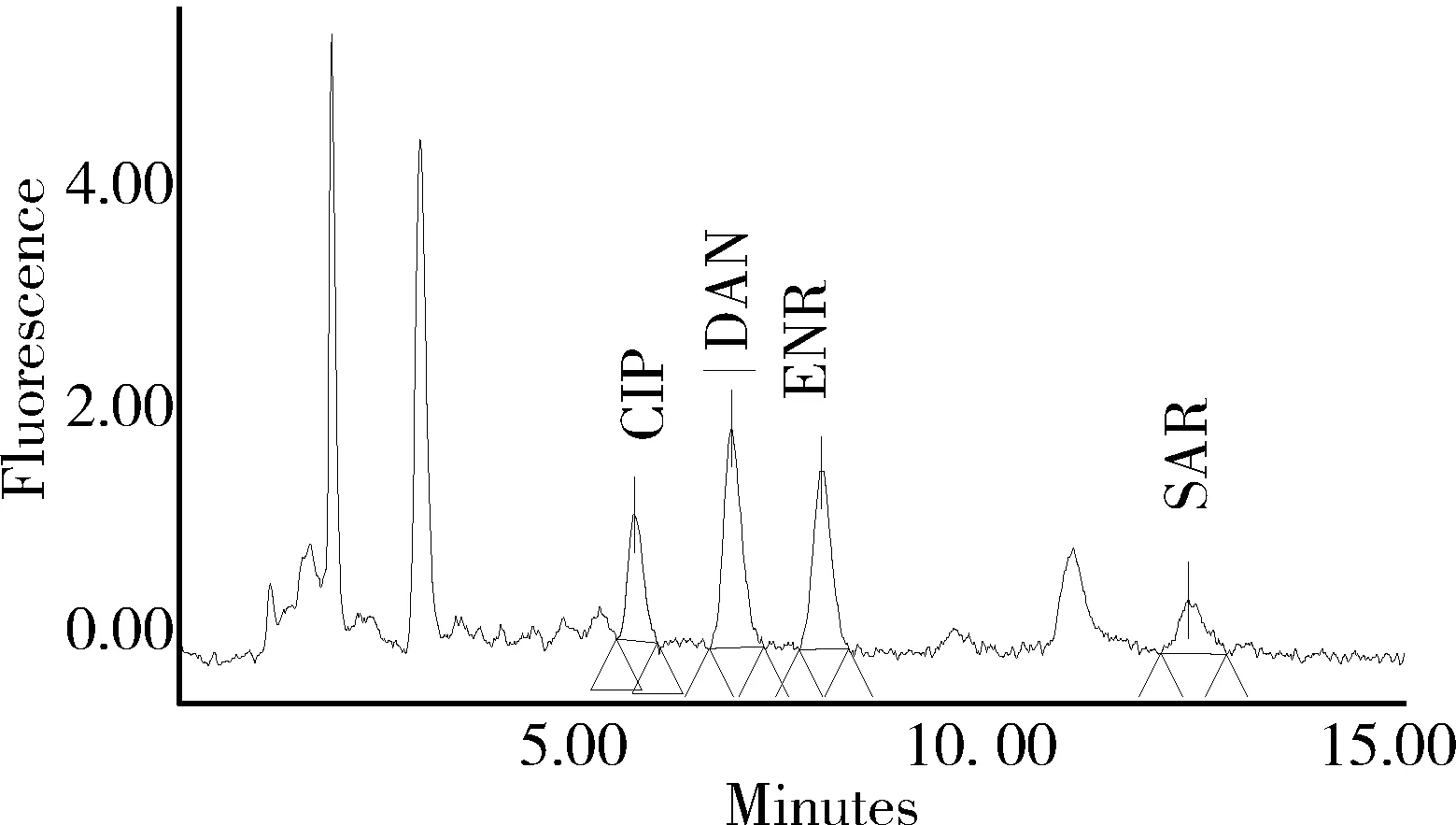
圖3 5 μg/kg空白雞蛋添加試樣中氟喹諾酮類藥物色譜圖Fig 3 Chromatogram of fluoroquinolones with 5 μg/kg added to blank egg
2.2 標準曲線 在選定的色譜條件下,配制系列標準工作液(1.4.3項)進行色譜分析,4種氟喹諾酮類藥物的回歸方程及相關系數見表1,標準曲線見圖4。

表1 4種氟喹諾酮類藥物測定的工作曲線Tab 1 Standard curves of 4 fluoroquinolones
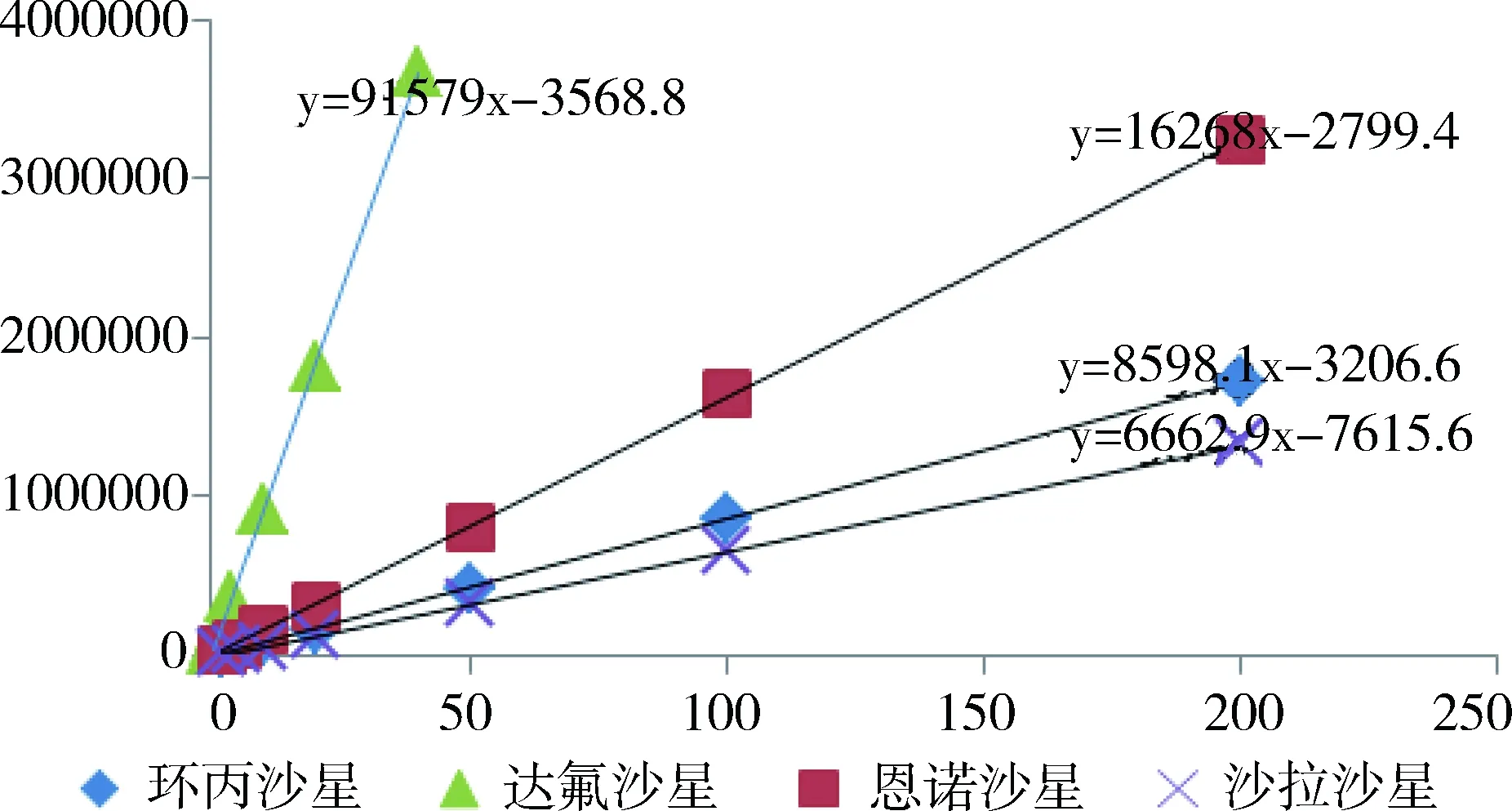
圖4 4種氟喹諾酮類藥物標準曲線Fig 4 Standard curves of 4 fluoroquinolones
2.3 回收率與精密度 用空白雞蛋添加后進行方法回收率試驗,結果見表2。由表2可見4種氟喹諾酮類藥物均具有良好的回收率和重現性。
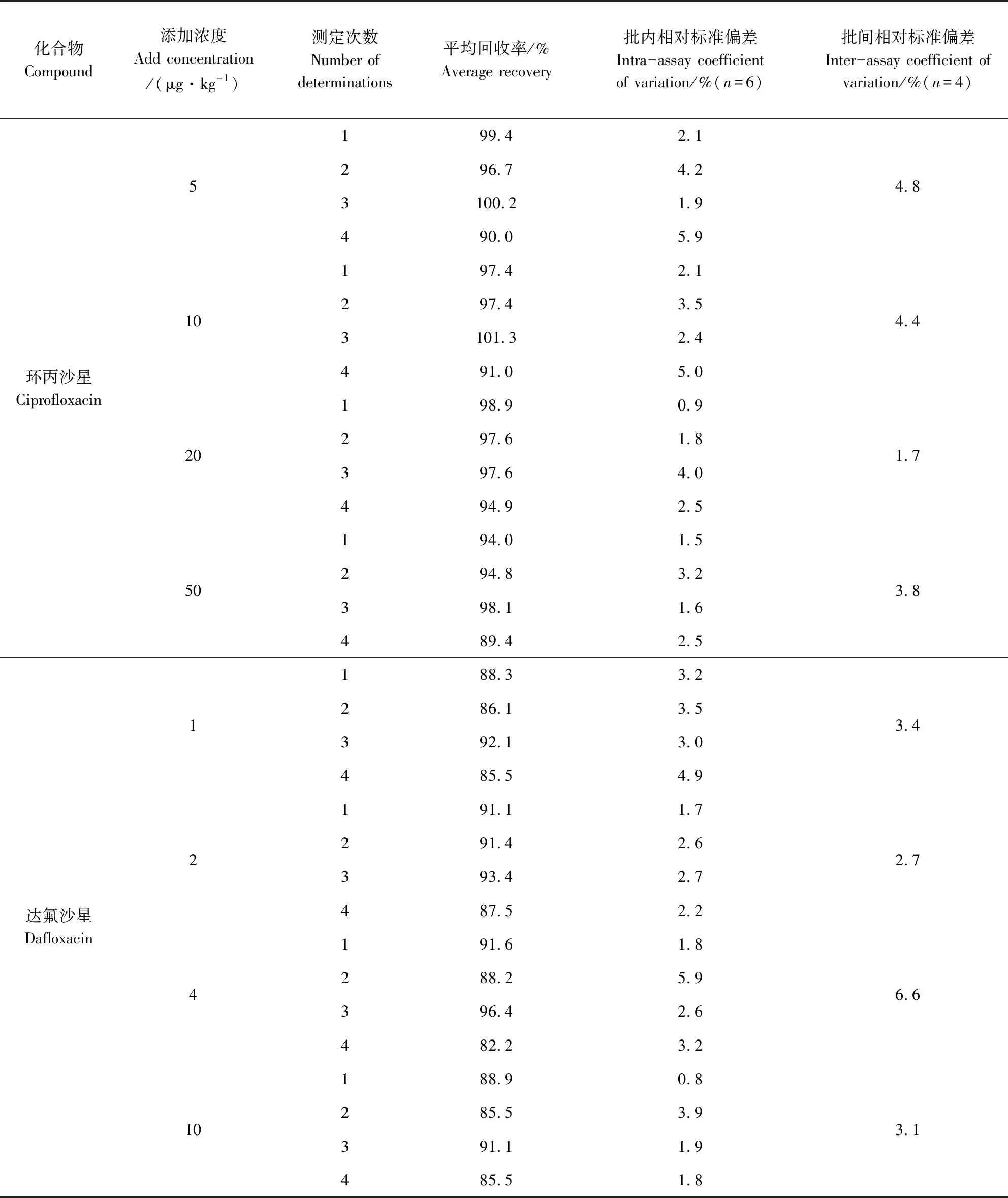
表2 雞蛋中氟喹諾酮類藥物殘留回收率試驗及批內批間相對標準偏差Tab 2 Recoveries of fluoroquinolone Remained in Eggs and the Intra-batch and Inter-batch RSDs
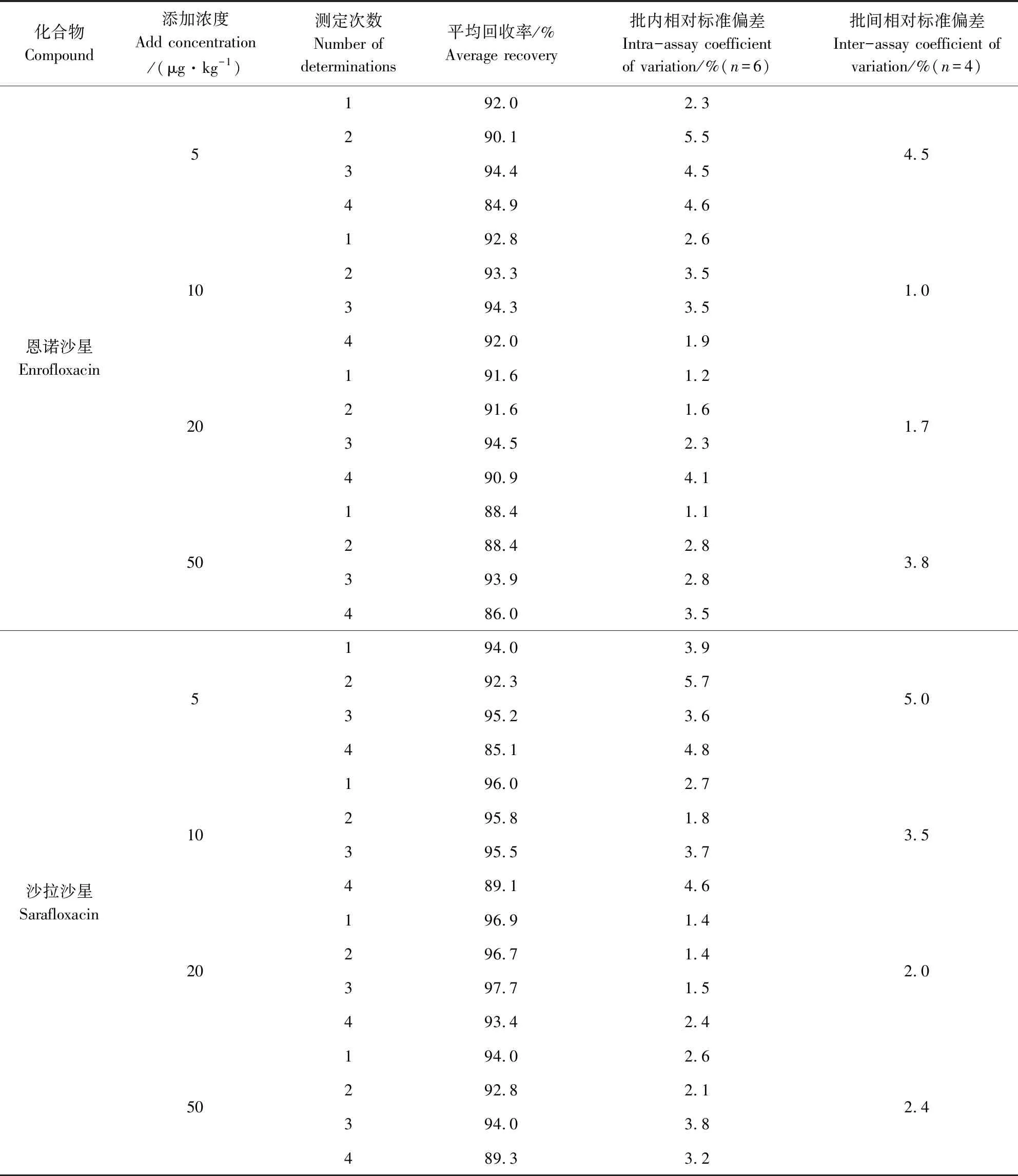
化合物Compound添加濃度Add concentration/(μg·kg-1)測定次數Number of determinations平均回收率/%Average recovery批內相對標準偏差Intra-assay coefficientof variation/%(n=6)批間相對標準偏差Inter-assay coefficient ofvariation/%(n=4)恩諾沙星Enrofloxacin5102050123412341234123492.090.194.484.992.893.394.392.091.691.694.590.988.488.493.986.02.35.54.54.62.63.53.51.91.21.62.34.11.12.82.83.54.51.01.73.8沙拉沙星Sarafloxacin5102050123412341234123494.092.395.285.196.095.895.589.196.996.797.793.494.092.894.089.33.95.73.64.82.71.83.74.61.41.41.52.42.62.13.83.25.03.52.02.4
2.4 檢測限 本方法的檢測限為環丙沙星、恩諾沙星、沙拉沙星5 μg/kg,達氟沙星1 μg/kg。
3 討論與結論
3.1 提取條件的選擇 目前,雞蛋中氟喹諾酮類藥物的提取液有乙腈[3]、甲酸乙腈溶液[8]、EDTA-Mcllvaine緩沖溶液[5]、磷酸鹽緩沖溶液[6-7]等。采用乙腈或酸化乙腈提取均需要增加濃縮步驟,較為繁瑣,而EDTA-Mcllvaine緩沖溶液的配制也較為復雜,故本實驗借鑒文獻[6-7]的提取方法,采用磷酸鹽緩沖溶液(pH 7.0)對雞蛋中氟喹諾酮類藥物進行提取。本實驗對振蕩時間、提取次數進行了考察,結果發現,樣品提取的振蕩時間大于10 min,4種氟喹諾酮類藥物的回收率無明顯變化。用磷酸鹽緩沖溶液(pH 7.0)提取2次比提取1次回收率提高9.8% ~15.6%,提取3次后,回收率無明顯增加。此外,由于雞蛋中含有大量水分,提取時與提取液混合在一起,用加入的提取液體積來計算結果會引起較大偏差,所以本實驗采用用磷酸鹽緩沖溶液(pH 7.0)提取2次,合并后定容的方法準確定量提取液體積。
3.2 脫脂條件的選擇 由于雞蛋中含有大量的脂肪、蛋白質及氨基酸等組分,樣品的提取液呈渾濁狀,高速離心無法使其澄清。所以,在固相萃取前應先進行脫脂。本實驗考察了不同體積、不同次數的正己烷脫脂效果,結果發現,用10 mL正己烷脫脂1次即能達到脫脂效果。需要注意的是,加入正己烷振蕩離心后正己烷和提取液會產生乳化,形成凝絮狀物質,低速離心往往使正己烷、乳化層、提取液層分層不明晰,采用高速(10000 r/min)離心10 min可改善分層現象。然而,用吸管不易吸取凝絮狀物質,所以,可用吸管小心吸取下層清液備用。
3.3 固相萃取條件的選擇 本實驗采用3 mL C18固相萃取柱凈化,比1 mL C18固相萃取柱能有效減少堵塞現象發生。首先用2 mL甲醇活化柱子,盡可能除去填料中存在的雜質,使填料溶劑化,提高固相萃取的重現性。再用2 mL磷酸鹽緩沖溶液平衡,為上樣創造一致的溶劑環境。本實驗比較了5、10、15、20 mL4種上樣體積的回收率,結果發現,5 mL和10 mL上樣量的回收率無明顯差異,但對于的低殘留樣品,采用5 mL上樣凈化時最終上機測試溶液的濃度較低,靈敏度不能滿足需要;而當上樣體積增加到15 mL以上后會出現不同程度的過柱緩慢甚至堵塞現象,回收率有所降低。所以,最終選擇取備用液10 mL進行凈化。之后用2 mL水淋洗固相萃取柱,除去其中的干擾物。淋洗完后,擠干固相萃取柱,盡可能除去柱中的殘留液體,再用2 mL 0.05 mol/L磷酸溶液/三乙胺(pH 2.4)-乙腈(82+18)即可完全洗脫。本實驗比較了不同體積、不同洗脫液組成比例的洗脫效果,結果發現,1 mL 0.05 mol/L磷酸溶液/三乙胺(pH 2.4)-乙腈(82+18)洗脫不完全,若增大乙腈比例,雖然可以提高洗脫效率但會引起色譜峰展寬。
3.4 方法比較 在空白雞蛋中添加環丙沙星、恩諾沙星、沙拉沙星20 μg/kg和達氟沙星4 μg/kg,采用該方法與常用標準進行比較,實驗結果見表3。
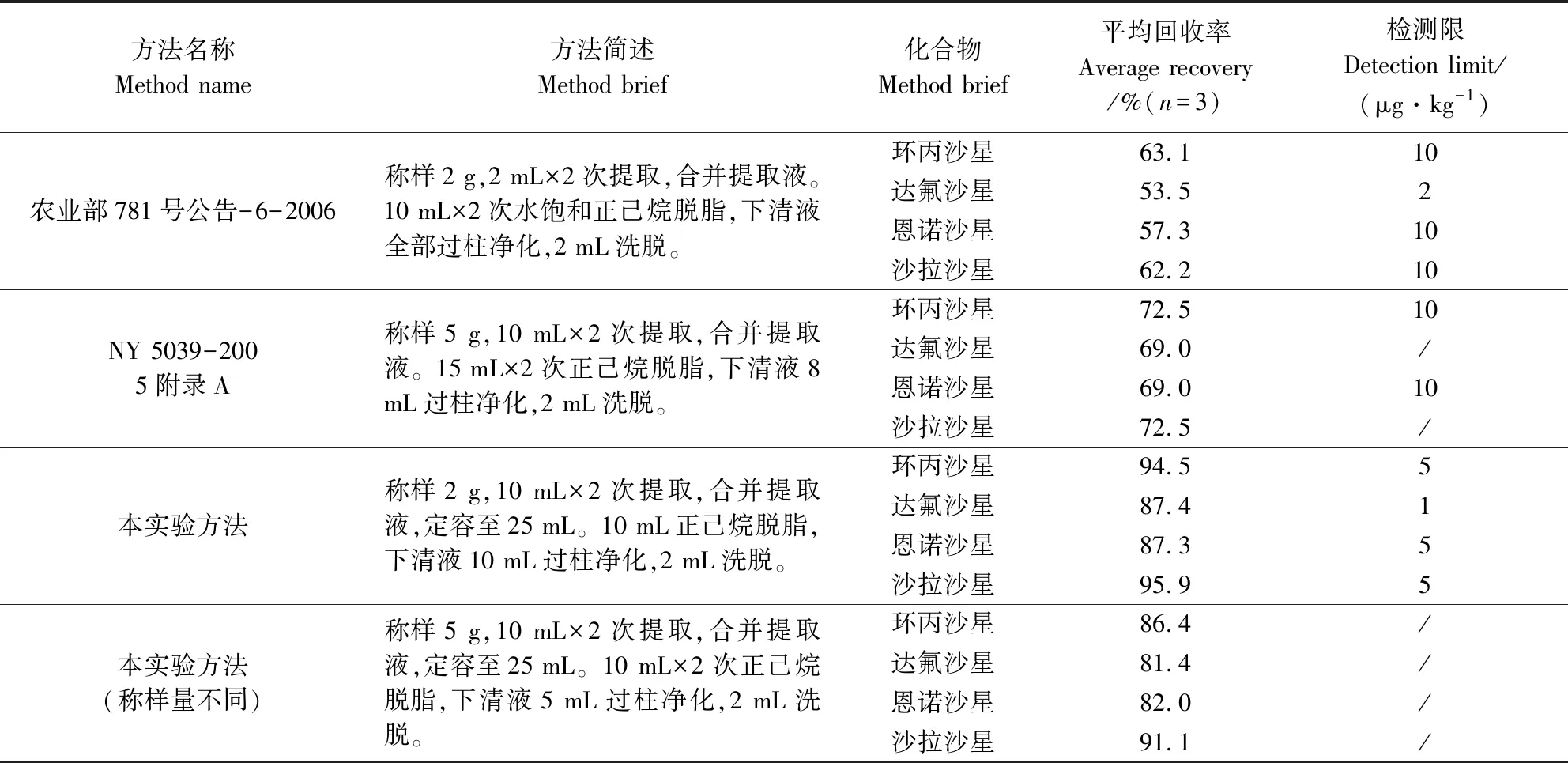
表3 不同方法測定雞蛋中氟喹諾酮類藥物殘留的比較Tab 3 Comparison of Different Methods for Testing of Fluoroquinolone Drugs Residues in Eggs
結果顯示:采用農業部781號公告-6-2006的檢測方法平均回收率低,原因在于合并提取液后脫脂時正己烷和提取液形成凝絮狀沉淀,除去沉淀后損失部分提取液,致使凈化液體積減少。按照公式A·Cs·V/As/m計算,凈化液與提取液體積不相等,樣品最終的稀釋體積產生偏差。而且,對于不同的樣品,視其脂肪等雜質含量的高低,形成的凝絮狀沉淀量不同,損失的提取液體積也不盡相同,其回收率的相對標準偏差也較大。
采用NY 5039-2005附錄A的方法回收率較低的主要原因是2次提取合并后雞蛋中本身的水分進入提取液,影響提取液的體積,且不同的樣品水分有所差別,致使提取液體積無法準確計量,按照公式A·Cs·V提取·V洗脫/As/m/V凈化計算,樣品提取液體積V1應為20 mL,實際值一般有24 mL左右,所以按加入的提取液體積計算的回收率結果會較低,且對于水分不同的樣品,它們之間的回收率的相對標準偏差也較大。
采用本實驗方法,平均回收率和相對標準偏差均滿意,提取后定容可以視為提取液體積為25 mL,脫脂后雖然提取液有損失,但取部分脫脂后的提取液凈化,其提取液、凈化液和定容液的體積均準確,按照公式A·Cs·V提取·V洗脫/As/m/V凈化計算,結果準確,回收率穩定,相對標準偏差小。將稱樣量增大至5 g時,平均回收率和相對標準偏差也均較滿意,但由于增加樣品的稱樣量,其脂肪等雜質的含量也相應提高,一般需要進行2次脫脂,增加了實驗步驟,故舍去該方法。
本研究通過優化前處理方法,建立起了液相色譜-熒光法同時檢測雞蛋中4種氟喹諾酮類藥物的方法,該方法線性范圍寬,靈敏度高,重復性好,可滿足日常樣品的檢測需求,完全適合雞蛋中氟喹諾酮類藥物殘留的檢測。
