高效液相色譜-串聯質譜法測定河蟹肝胰腺組織中林可酰胺類抗生素殘留
2022-01-07 02:27:02邱稀木朱曉華邊文冀王淑芳
福建農業學報 2021年10期
邱稀木,朱曉華,邊文冀,王淑芳,孟 勇
[1. 江蘇海洋大學海洋科學與水產學院,江蘇 連云港 222005;2. 江蘇省淡水水產研究所,江蘇 南京 210017;3. 江蘇省水產質量檢測中心/農業農村部漁業產品質量監督檢驗測試中心(南京),江蘇 南京 210017]
0 引言
【研究意義】林可酰胺(Lincosamide)類抗生素主要有林可霉素(Lincomycin)、克林霉素(Clindamycin)、吡利霉素(Pirlimycin),能抑制細菌的蛋白質合成,對大多數革蘭氏陽性菌和一些厭氧革蘭氏陰性菌有抗菌作用[1-2]。林可酰胺類抗生素主要運用于臨床和畜禽養殖上,根據農業農村部印發的《水產養殖用藥明白紙2020年1、2號》,林可酰胺類抗生素藥物在水產養殖上的運用目前還沒有明確的指導屬于水產養殖的非規范用藥。近年來,在漁用投入品的風險篩查中,在某些改水類產品中檢測到林可酰胺類抗生素藥物成分,對水產養殖帶來了一定的風險隱患。【前人研究進展】目前,關于林可酰胺類藥物殘留的檢測方法比較多,如液相色譜-串聯質譜法測定蜂蜜中的林可酰胺類獸藥殘留[3]、酶聯免疫分析方法測定牛肉雞肉豬肉魚肉中的林可酰胺類抗生素[4]、高效液相色譜法對林可霉素含量的測定[5]、林可霉素的磁免疫化學發光檢測試劑盒及其應用[6]等,但在河蟹樣品中林可酰胺類藥物的檢測方法還未見報道。其中,液相色譜法以保留時間作為定性標準,比較容易產生假陽性的結果,而且靈敏度不高[7];化學發光法需要柱后衍生,操作的過程較為繁瑣,時間和人工成本高[7]。且河蟹的肝胰腺樣品油脂成分高存在基質效應,基質常常對分析物的分析過程有顯著的干擾,并影響分析結果的準確性,所以對樣品提取和凈化的方法要求會較高[8]。【本研究切入點】固相萃取(SPE小柱)主要用于從各類復雜的基質提取出想得到的分析物,例如生物體(血液和尿液)、水樣[9]、土壤、藥材[10]中提取目標分析物,達到濃縮或純化的目的。本研究將固相萃取小柱用于河蟹肝胰腺樣品的前處理,以期達到更好的提取和凈化效果,降低其他復雜基質對樣品提取的影響。【擬解決的關鍵問題】本研究以3種林可酰胺類抗生素(林可霉素、克林霉素、吡利霉素)為檢測對象,將SPE固相萃取柱應用于河蟹的獸藥殘留檢測前處理,以期建立河蟹中林可酰胺類抗生素獸藥殘留的快速檢測方法,對河蟹質量安全管控具有積極的意義。
1 材料與方法
1.1 儀器和試材料
1.1.1 儀器 QTRAP 4500三重四極桿(美國SCIEX公司,配備電噴霧離子源);實驗室用水(Millipore系統超純水);漩渦振蕩器(北京佳源興業公司);Allegra TM 離心機(美國Beckman公司);Turbo Vap濃縮工作站(瑞典Biotage公司)。
1.1.2 材料 甲醇、乙腈(色譜純,德國Merck公司);甲酸(色譜純,美國ROE公司);有機濾膜(0.22 μm,天津津騰有限公司);OASIS?HLB 6 :500(mL:mg)固相萃取柱;3種林可酰胺類抗生素標準品(農業農村部環境保護科學研究監測所,100 μg·mL-1);河蟹樣品(從南京各農貿市場隨機抽樣)。
1.2 標準溶液的制備
準確稱取5.0 mL林可酰胺、克林霉素和吡利霉素標準品溶液,分別轉移至同一50.0 mL容量瓶中,用乙腈作為稀釋劑溶解并稀釋至一定體積。進一步將5.0 mL該溶液轉移至50.0 mL容量瓶中,用乙腈稀釋劑稀釋至一定體積。
1.3 樣品的提取和凈化
1.3.1 樣品提取 進行以下步驟:(1)將5 g河蟹樣品(蟹肉和蟹黃混合)稱量到離心管中;(2)加入配置好的2%甲酸乙腈15 mL;(3)使用漩渦振蕩器渦旋5分鐘使提取劑與樣品充分溶解;(4)將每管樣品在4 ℃下以9000 r·min-1的速度離心10 min;(5)離心后的上清液轉移到另一個50 mL離心管中;(6)殘留物用10 mL提取液再次提取,合并上清液。(7)溶液中加入5 mL正己烷,渦旋振蕩30 s,9000 r·min-1的速度離心5 min,棄去 上層正己烷,再加入5 mL正己烷重復操作一次。下層溶液待凈化。
1.3.2 樣品凈化 SPE小柱依次用6 ml甲醇和6 ml超純水以3 mL·min-1的流速進行預處理。將上清液(10 mL)以1 mL·min-1的速度通過固相萃取柱,棄流出液。10 mL超純水沖洗柱子,棄流出液,抽真空10 min。5 mL甲醇沖洗柱子洗脫。在40 ℃的條件下,洗脫液用氮吹儀吹至完全干,氮吹干后的殘留物加入1.00 mL 0.1%甲酸水-甲醇(9∶1)混合溶液,2分鐘渦旋使其充分混合,用針管吸取混合溶液,將溶液通過0.22 μm有機濾膜,置入棕色樣品瓶中,待上機。
1.4 色譜和質譜分析條件
1.4.1 液相色譜條件 色譜柱:InfinityLab poroshell 120 SB-C18,(100 mm×2.7 mm,2.7 μm);A相為0.1%甲酸水溶液,B相為甲醇;柱溫:30 ℃;樣品量:10 μL;流速:0.40 mL·min-1;梯度洗脫程序見表1。

表1 流動相梯度洗脫程序Table 1 Procedures of mobile phase gradient elution
1.4.2 質譜分析條件 質譜的電離模式為電噴霧離子源(ESI+模式);輔助加熱氣體流量:50 L·min-1;霧化氣體流量:50 L·min-1;離子源的溫度為550 ℃;氣簾氣流量:35 L·min-1;噴霧電壓設置為5500 V。
2 結果與分析
2.1 提取溶劑優化
林可酰胺類抗生素極性較強,容易溶于極性溶劑[2,11]。因此,比較了磷酸鹽緩沖液、乙腈、乙酸乙酯和丙酮的萃取效率。結果表明,磷酸鹽緩沖液含水量高,不利于后續的濃縮,溶解的蛋白易導致過SPE柱時堵塞;乙酸乙酯作為萃取溶劑時,乳化現象嚴重,回收率很低;丙酮用作提取溶劑時揮發性很高,直接注入可能會破壞管道和質譜組件[3]。乙腈是極性較強的有機溶劑,具有沉淀蛋白質的作用,各指標的回收率優于其他提取劑。因此,本試驗選擇乙腈為提取溶劑。此外,分別以純乙腈、1%甲酸乙腈、2%甲酸乙腈、1%氨水乙腈、2%氨水乙腈作為提取液,考察其對3種林可酰胺類目標物的提取效果。結果顯示,相比其他組,2%甲酸乙腈作為提取溶劑時的平均回收率最高,3種林可酰胺類藥物的回收率為76.9%~102%,滿足期望結果回收率在75%以上。因此,本試驗提取樣品采用2%甲酸乙腈進行試驗。
2.2 色譜條件的優化
利用InfinityLab poroshell 120 SB-C18(100 mm×2.7 mm,2.7 μm)色譜柱在ESI+模式下,比較了在乙腈-水或甲醇-水流動相系統中3種林可酰胺類藥物的色譜結果。試驗結果是:甲醇-水流動相中3個目標的峰值形狀和響應優于乙腈-水,且洗脫時間短,所以選擇了甲醇-水流動相系統。
通過在流動相中加入適量的揮發性鹽或酸可以提高目標物的反應值、分辨率和峰形[12-13]。因此,在水相中添加甲酸或乙酸銨對流動相系統做進一步優化。離子化后3種目標化合物主要以[M+H]+的形式存在[14-15]。結果是,在水相中添加醋酸銨降低了各目標化合物的響應值[16-18];在水相中加入甲酸后,各目標化合物的質譜響應提高了10%,且添加0.1%甲酸時效果最佳。因此本試驗使用0.1%甲酸水-甲醇作為流動相。
2.3 質譜參數的優化
將50 μg·L-1待測林可酰胺標準混合溶液用針型泵注入質譜儀,分別用電噴霧正電離模式和電噴霧負電離模式對藥物的母離子全掃描進行比較并優化了碰撞能量和去簇電壓。分析結果表明,在電噴霧正電離模式下,3種測試對象的離子信號穩定,響應速度快,因此選擇電噴霧正電離掃描模式。在這種模式下,所有3種化合物都產生了[M+H]+的母離子。進一步掃描生成的子離子,選擇豐度最高的2個子離子進行定性和定量分析。考察了噴霧電壓分別為:4500V、5000 V、5500 V時,3種標準溶液的離子化效率,結果為5500 V時的離子化效率最高。目標化合物的質譜分析條件見表2。3種林可酰胺類抗生素藥物的總離子流圖見圖1。

圖1 質量濃度為50 ng·L-1的 3 種林可酰胺類藥物標準溶液的總離子流圖Fig.1 Total ion chromatograms of 50 ng·L-1standard solutions of 3 lincosamide antibiotics

表2 林可酰胺類藥物測定的質譜參數Table 2 Mass spectrometric parameters for lincosamide antibiotics determination
2.4 SPE小柱對蟹黃的凈化效果
河蟹的肝胰腺富含碳水化合物、氨基酸、有機酸、蛋白質、膠原蛋白、維生素A、不飽和脂肪酸等,這會影響目標化合物的測定,并污染離子源[19-21]。因此,本試驗考察了在河蟹肝胰腺樣品提取中,固相萃取柱對3種林可酰胺類藥物的凈化效果和回收率。試驗分析了6種SPE小柱的凈化效果(圖2),其中OASIS?HLB 6 mL/500 mg SPE柱組的凈化結果最好。使用該型號SPE小柱,3種林可酰胺類藥物的回收率在80%以上,平均回收率是84.6%,明顯好于其他固相萃取柱組。因此,本方法最終選擇型號為OASIS?HLB 6 mL/500 mg的 SPE柱來凈化河蟹肝胰腺樣品。

圖2 不同固相萃取柱對目標物回收率的影響Fig. 2 Recovery rates on 3 antibiotics by various SPE columns
2.5 基質效應的考察
電噴霧離子源在電離時候,樣品中的基質和目標化合物具有競爭關系,這種現象為基質強化效應或基質抑制效應[22-24]。本試驗考察了1、5、10、20、50、100 ng·mL-16個質量濃度,建立對應的直角坐標軸,峰面積為縱坐標,質量濃度為橫坐標。基質效應(ME)的計算公式為(基質標準工作曲線的斜率/溶劑標準工作曲線的斜率)×100%[25-27]。使用6種固相萃取柱進行二次純化后,基質工作曲線與標準工作曲線的斜率之比均低于50%,表明具有顯著的基質效應。因此,本方法采用基質匹配曲線對 3 種林可酰胺類抗生素獸藥殘留進行定量分析。相比于其他組,OASIS?HLB 6 :500(mL:mg)固相萃取小柱可以有效降低河蟹肝胰腺樣品在液相色譜質譜聯用儀上的基質抑制效應。
2.6 試驗方法的評價
2.6.1 方法的線性范圍、檢測限和定量限 以0、1、5、10、20、50 ng·mL-1為質量濃度水平,以峰面積Y和相應的目標物質質量濃度為X(μg·L-1),繪制出3種林可酰胺類抗生素的基質標準工作曲線,線性相關系數(R2)均高于0.99,線性回歸方程和相關系數見表3。將含有3種目標物質的標準溶液添加到空白河蟹樣品中,制備具有不同目標物質含量的加標樣品。根據優化條件測定樣品,分別采用3倍和10倍信噪比(S/N)的計算方法來確定方法的檢測限度和定量限度[28-29]。本檢測方法目標物的LOD值約為0.3 μg·kg-1,LOQ值約為1.0 μg·kg-1(表3)。本方法的線性范圍能夠滿足實際河蟹肝胰腺樣品的檢測需要。
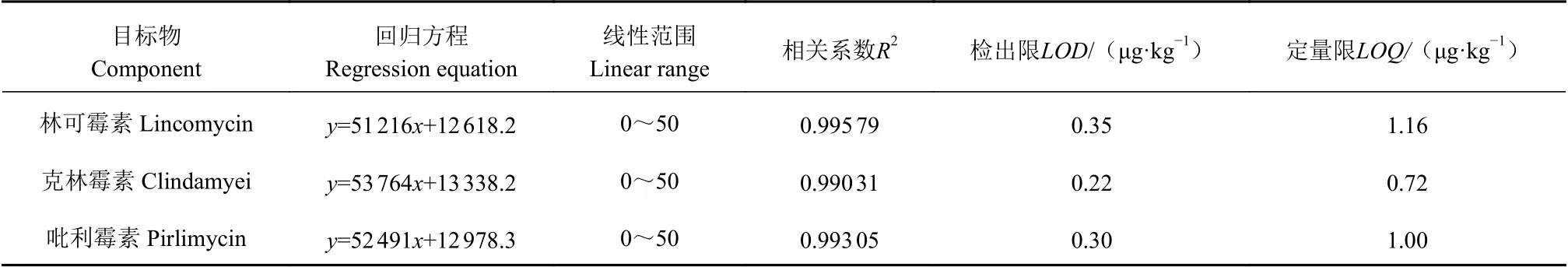
表3 方法的線性回歸方程、相關系數、檢出限和定量限Table 3 Linear regression equation, correlation coefficient, detection limit, and quantitative limit of assay
2.6.2 方法的準確性和精密度 將混合標準溶液添加到河蟹的空白樣品中,以獲得目標質量濃度為5.0、10.0和50.0 μg·kg-1的3個加標水平,并根據優化的方法重復測定6次。河蟹樣品中3種林可酰胺類抗生素的加標回收率和精度見表4。3種質量濃度的林可酰胺類抗生素在蟹類樣品中的回收率為80.5%~99%,相對標準差(RSD)為2.5%~5.2%,回收率和準確度高,能夠滿足河蟹樣品中這3種林可酰胺類抗生素殘留的分析要求。

表4 3種林可酰胺類藥物的加標回收率及精密度(n=6)Table 4 Recovery and precision of assay on 3 lincosamide antibiotics (n=6)
2.7 實際樣品分析
用本方法檢測了從各農貿市場抽查到的170份河蟹樣品。結果5份河蟹樣品林可霉素含量檢出。檢出的陽性樣品林可霉素質量濃度依次為:4.43 、9.23、4.26 、17.77 、8.47 μg·kg-1。本方法簡單、快捷、精確度高,適用于河蟹樣品中3種林肯酰胺類抗生素殘留的檢測,其中一份陽性樣品的色譜圖如圖3所示。

圖3 陽性樣品的色譜圖Fig. 3 Chromatograms of positive samples
3 討論與結論
復雜的油脂基質給河蟹蟹黃抗生素類藥物的檢測造成了一定的困難。油脂易溶于正己烷,正己烷和水分層,正己烷在上層,棄掉正己烷可達到凈化效果;HLB小柱吸附劑是由親脂性二乙烯苯和親水性n-乙烯基吡咯烷酮兩種單體按一定比例聚合成的大孔共聚物,其保留機理為反相,通過一個“特殊的極性捕獲基團”來增加對極性物質的保留提供很好的水浸潤性,林可酰胺類抗生素藥物是極性較強的物質,因此用HLB小柱提取該藥物。
針對河蟹肝胰腺基質的特點,結合現行有效標準和文獻報道,本研究對傳統提取工藝進行了改進,以減少肝胰腺樣品中其他復雜基質的干擾,使用2%甲酸乙腈作樣品的提取溶劑,凈化過程采用了正 己 烷 祛 脂 和OASIS?HLB 6∶500(mL∶mg)SPE柱進行過濾和富集,優化了部分液相色譜和質譜的分析程序等。采用高效液相色譜-質譜聯用技術,建立了在河蟹肝胰腺樣品中林可酰胺類抗生素藥物殘留的快速測定方法。本方法具有步驟少、精確度高、穩定性好、重現性好、減少溶劑的消耗量等優點。本研究完善了對河蟹肝胰腺樣品中林可酰胺類獸藥殘留的定量檢測,可為河蟹的質量安全生產監控提供技術支持。
