UHILIC-MS/MS同時測定栝樓桂枝顆粒中22個氨基酸的含量
2022-02-28 09:16:24蔣昆霞朱美玲王雅心陳小婷林羽許文徐偉
藥學研究 2022年1期
蔣昆霞,朱美玲,王雅心,陳小婷,林羽,許文*,徐偉*
(1.福建中醫藥大學藥學院,福建 福州 350122;2.福建中醫藥大學生物醫藥研發中心,福建 福州 350122)
栝樓桂枝湯源自張仲景的《金匱要略》,福建中醫藥大學陳立典教授結合多年的臨床經驗,將栝樓桂枝湯應用于腦卒中后的康復取得良好的療效,目前該復方的顆粒劑栝樓桂枝顆粒經院內制劑新藥申報批準為福建省第二人民醫院制劑(批件號:閩2013S0001),主要用于腦卒中后肢體痙攣及康復治療[1-2]。現代藥理研究表明,還可用于治療缺血性腦卒中、癲癇、脊髓損傷后的肢肉痙攣等[3-5]。
現代藥理學研究表明栝樓桂枝顆粒可以通過調控神經抑制性氨基酸——γ-氨基丁酸(GABA)受體發揮改善腦卒中后痙攣作用[6],也可以調節興奮性氨基酸及其受體的表達,改善MCAO大鼠模型大鼠的神經損傷[7],代謝組學研究顯示栝樓桂枝湯可以改善體內氨基酸代謝異常[8]。提示栝樓桂枝顆粒中氨基酸類成分可能與其在體內調控神經調控相關的氨基酸受體有關,尤其君藥栝樓根富含多種氨基酸類化合物[9]。
目前復方中關于氨基酸類化合物的分析方法主要采用高效液相色譜(HPLC)法[10-13],由于氨基酸屬于強極性成分,在普通反相色譜上往往難以保留,常規HPLC方法的樣品前處理相對較復雜,往往需要進行柱前衍生,但是分析測定時容易受到衍生劑的穩定性以及衍生速度的影響,導致測定方法的靈敏度不高,難以滿足對中藥復方樣品進行快速、高靈敏度和高通量的分析要求。近幾年,隨著現代色譜-質譜分析技術的發展,超高效親水作用色譜,越來越多的用于中藥復方中強極性成分的分離,通過串聯質譜可高效的應用于中藥復方中復雜體系的分析[14-15]。因此,本文通過建立一種超高效親水作用色譜串聯三重四極桿質譜法(UHILIC-MS/MS)對栝樓桂枝顆粒中22種氨基酸類成分進行無衍生化的快速含量測定,從氨基酸角度探討栝樓桂枝顆粒抗腦卒中發揮神經保護作用的藥效物質基礎提供實驗依據。
1 儀器與試藥
1.1 儀器 ACQUITY UPLC I-Class超高效液相色譜儀(美國Waters公司);Xevo TQS三重四極桿質譜(美國Waters公司);CPA225D型十萬分之一分析天平(德國Sartorius公司);Milli-Q超純水儀(美國Millipore公司)。
1.2 試藥 甲醇、乙腈(質譜純,德國Merk公司),甲酸(色譜純,批號:F190210,阿拉丁試劑上海有限公司),其余試劑均為分析純。亮氨酸(Leu)、苯丙氨酸(Phe)、色氨酸(Trp)、異亮氨酸(Ile)、蛋氨酸(Met)、脯氨酸(Pro)、酪氨酸(Tyr)、丙氨酸(Ala)、蘇氨酸(Thr)、甘氨酸(Gly)、谷氨酸(Glu)、絲氨酸(Ser)、精氨酸(Arg)、組氨酸(Arg)、賴氨酸(Lys)、纈氨酸(Val),以上對照品均購自中國食品藥品檢定研究院,批號:140681-201703;γ-氨基丁酸(GABA,A104200)、羥脯氨酸(Hpro,H111005)、天冬酰胺(Asn,A105949)、門冬氨酸(Asp,I1412118)、瓜氨酸(Cit,C109228)、谷氨酰胺(Gln,G103978)均購自上海阿拉丁生化科技股份有限公司;氘代甘氨酸(D-Gly,TRC1599)購于加拿大Toronto Research Chemicals Inc公司,純度均大于98%。栝樓桂枝顆粒來自福建省第二人民醫院院內制劑。
2 方法與結果
2.1 色質譜條件
2.1.1 色譜條件 Waters ACQUITY UPLC BEH Amide (2.1 mm×100 mm,1.7 μm),流動相:(0.25%甲酸+2 mmol·L-1甲酸銨)水(A)-乙腈(B),親水作用色譜梯度洗脫程序:0~2 min,8%A→8%A;2~3 min,8%A→35%A;3~4 min,35%A→45%A;4~4.1 min,45%A→8%A;4.1~6 min,8%A,進樣量:1 μL,流速:0.4 mL·min-1,柱溫:45 ℃。
2.1.2 質譜條件 電噴霧正離子模式;毛細管電壓2.5 kV;脫溶劑氣流:氮氣800 L·h-1;脫溶劑溫度:200 ℃;錐孔氣流:氮氣50 L·h-1;離子源溫度:150 ℃;Extractor:3.00 V;碰撞氣體:氬氣。
2.2 溶液的制備
2.2.1 對照品溶液 取Leu、Phe、Trp、Ile、GABA、Met、Val、Pro、Tyr、Ala、Hpro、Thr、Gly、Glu、Gln、Ser、Asn、Asp、Cit、Arg、Hit、Lys的對照品適量,精密稱定,加入50%甲醇分別制備質量濃度為148.5、130.25、221.5、89.8、693.0、5.93、340.5、1 584、178.2、244.8、5.12、257.5、256.0、275.5、51.3、128.5、1 110、406.0、1 784、564、352.0、84.0 μg·mL-1的單一對照品儲備液。分別精密量取上述對照品儲備液適量至100 mL量瓶中,加入50%甲醇定容,即得以上成分的質量濃度分別為1 485、1 302.5、2 215、898、6 930、59.3、3 405、15 840、1 782、2 448、51.2、2 575、2 560、2 755、513、1 285、11 100、4 060、17 840、5 640、3 520、840 ng·mL-1的混合對照品溶液。
2.2.2 內標溶液 精密稱定對照品氘代甘氨酸(D-Gly)適量,加入甲醇溶解并稀釋至刻度,制備質量濃度為1.03 mg·mL-1的單一對照品儲備液。精密量取上述對照品儲備液適量至10 mL量瓶中,加入50%甲醇定容,即得質量濃度為1.03 μg·mL-1的內標溶液。
2.2.3 供試品溶液 取栝樓桂枝顆粒約0.5 g,精密稱定,置具塞三角瓶中,精密加入50%甲醇100 mL,密塞,稱量,超聲處理(功率250 W,頻率40 kHz)30 min,放冷,再稱量,用超純水補足失重,搖勻,0.22 μm濾膜濾過,取續濾液即得。用于定量分析的樣品溶液:按1∶1(200,200 μL)加入內標溶液后進行測定。
2.3 方法學考察
2.3.1 質譜測定方法 采用正離子多反應監測(MRM)定量模式,22種分析物及內標的質譜分析條件參數見表1。混合對照品和栝樓桂枝顆粒樣品(S1)在“2.1”項色譜及質譜條件下分析得栝樓桂枝顆粒的UHILIC-MS/MS的MRM色譜圖(見圖2)。結果表明22種分析物對照品與栝樓桂枝顆粒中所鑒定成分的色譜峰保留時間符合定性定量分析要求。
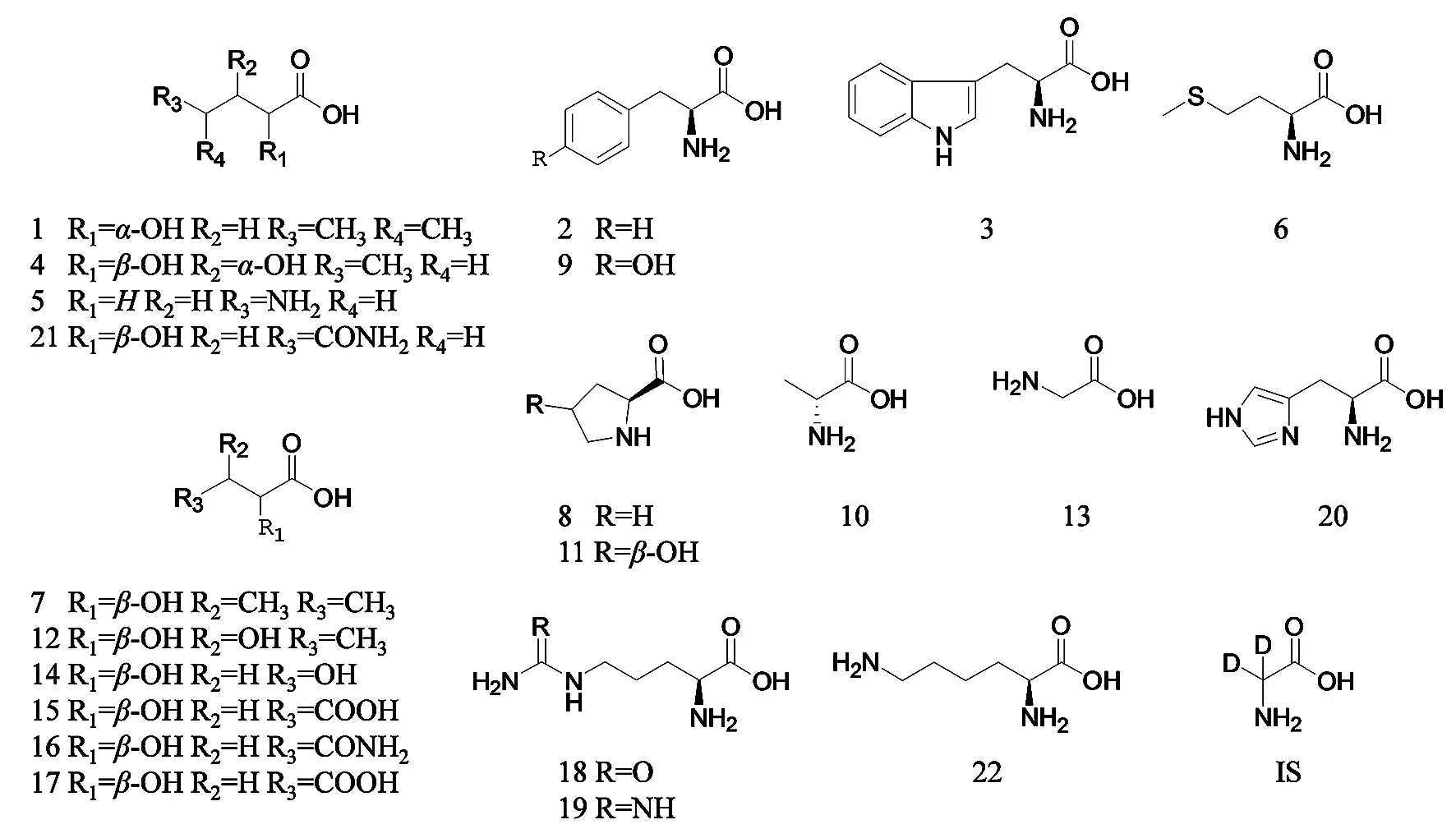
圖1 各成分及內標物化學結構圖1.Leu;2.Phe;3.Trp;4.Ile;5.GABA;6.Met;7.Val;8.Pro;9.Tyr;10.Ala;11.Hpro;12.Thr;13.Gly;14.Glu;15.Gln;16.Ser;17.Asn;18.Asp;19.Cit;20.Arg;21.Hit;22.Lys;IS.D-Gly
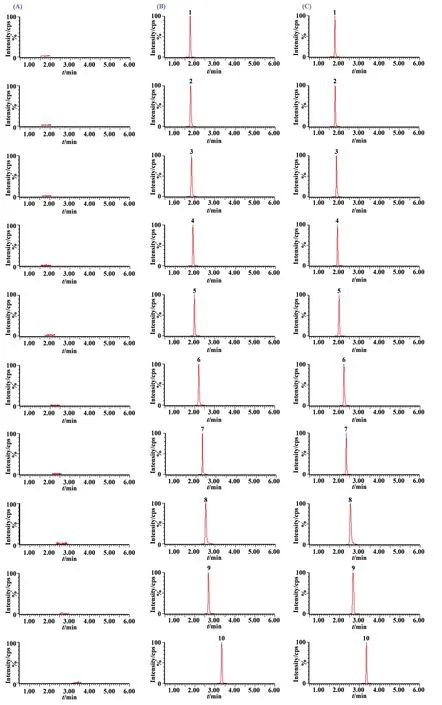
圖2 空白溶液(A)、對照品(B)和栝樓桂枝顆粒樣品(C)的UHILIC-MS/MS色譜圖1.Leu;2.Phe;3.Trp;4.Ile;5.GABA;6.Met;7.Val;8.Pro;9.Tyr;10.Ala;11.Hpro;12.Thr;13.Gly;14.Glu;15.Gln;16.Ser;17.Asn;18.Asp;19.Cit;20.Arg;21.Hit;22.Lys;IS.D-Gly
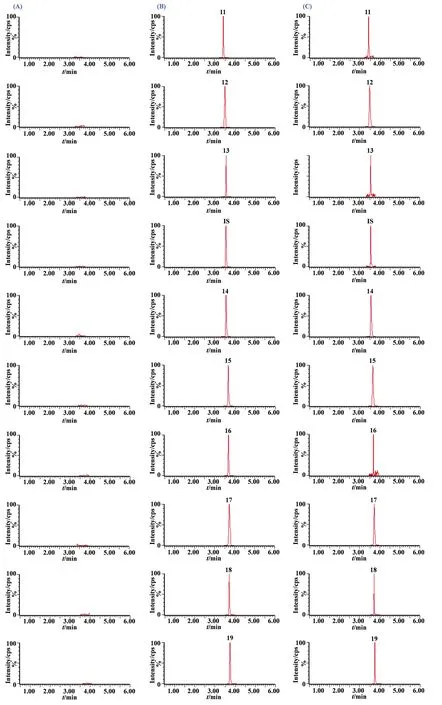
圖2(續)
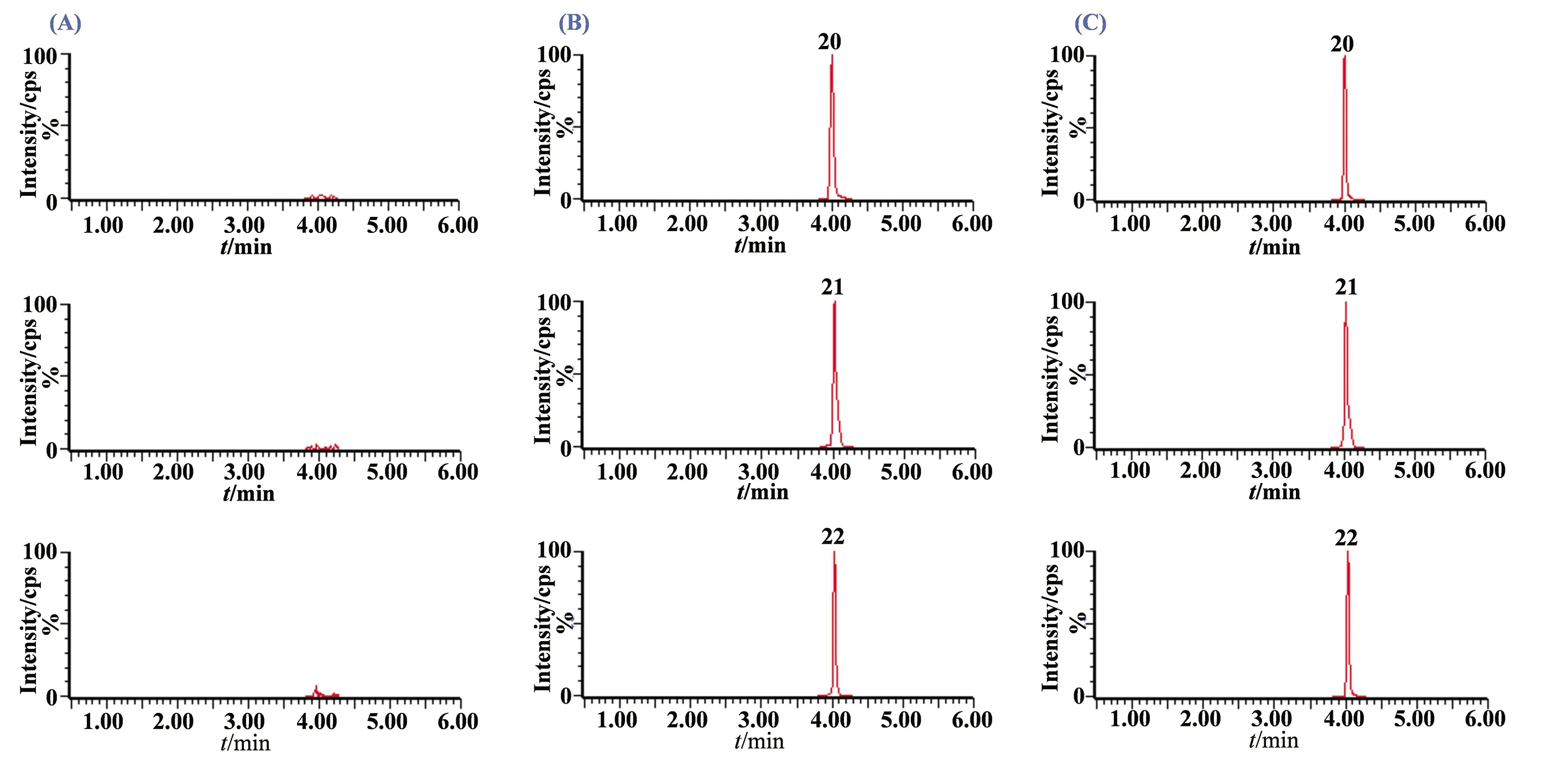
圖2(續)
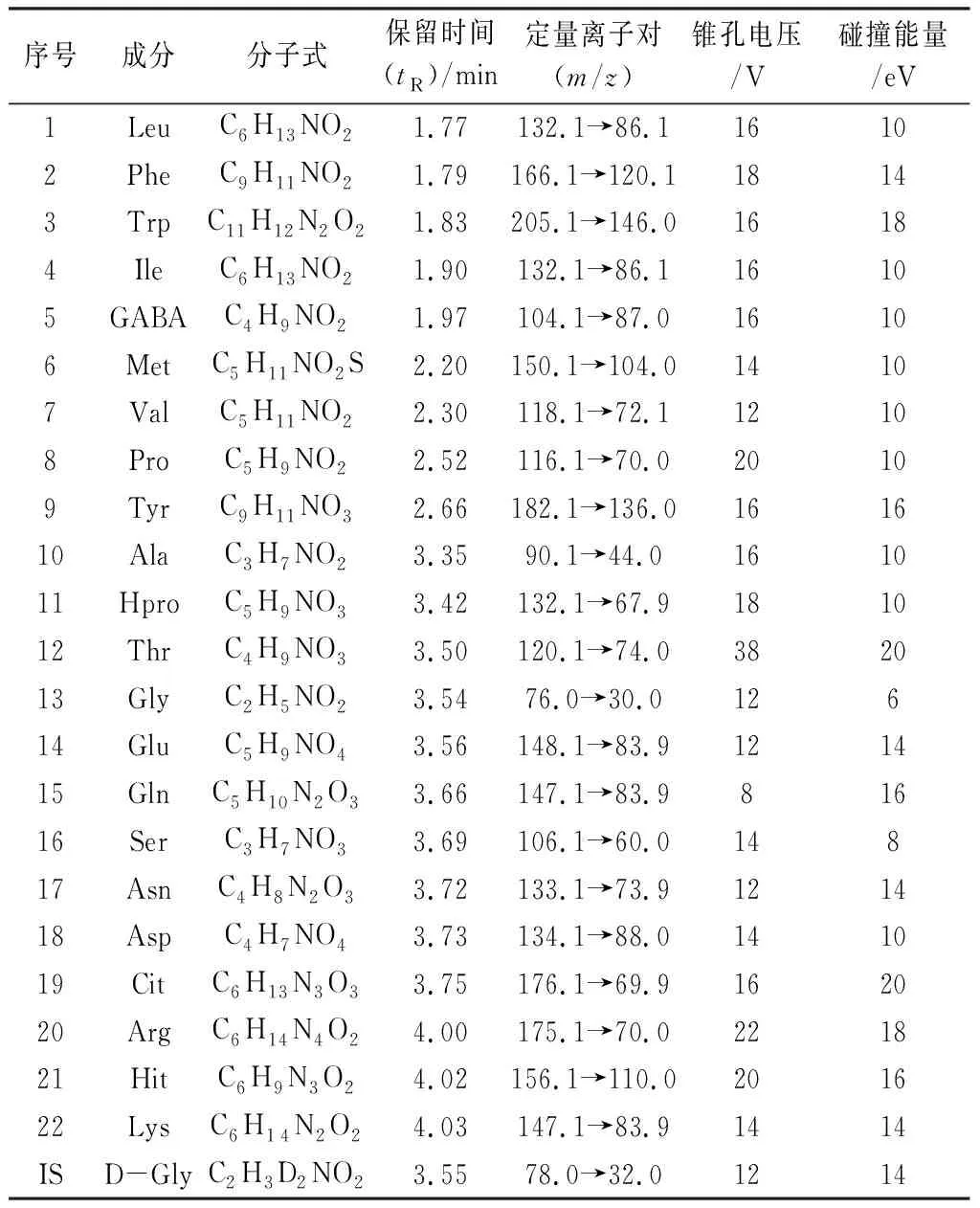
表1 22個氨基酸及內標的質譜測定條件
2.3.2 線性和范圍 取“2.2.1”項下方法制備的混合對照品溶液,用50%甲醇逐級稀釋1、2、5、10、25、50倍系列梯度濃度的對照品混合溶液。按“2.1”項下條件測定峰面積,用峰面積與內標峰面積的比值(Y)對分析物質量濃度(X)進行線性回歸,繪制標準曲線,得到回歸方程和相關系數,結果見表2。

表2 22種氨基酸的回歸方程、線性范圍以及相關系數
2.3.3 精密度試驗 取“2.2.1”項下混合對照品溶液,按“2.1”項條件1 d內連續進樣6次及連續3 d重復進樣(每天重復進樣3次)測定,測定各成分和內標的峰面積,分別計算其峰面積比的RSD,其日內精密度和日間精密度結果見表3,表明儀器精密度良好。
2.3.4 穩定性試驗 按“2.2.3”項下方法制備供試品溶液(S1批次),分別于0、2、6、10、12和24 h按“2.1”項下條件進樣測定,測定各成分和內標的峰面積,求出峰面積比的RSD。結果見表3,表明供試品溶液在24 h內穩定。
2.3.5 重復性試驗 精密稱取同一批栝樓桂枝顆粒6份(S1批次),按“2.1”項下條件進樣測定,計算22個分析物的含量及其RSD,結果見表3,表明方法重復性良好。
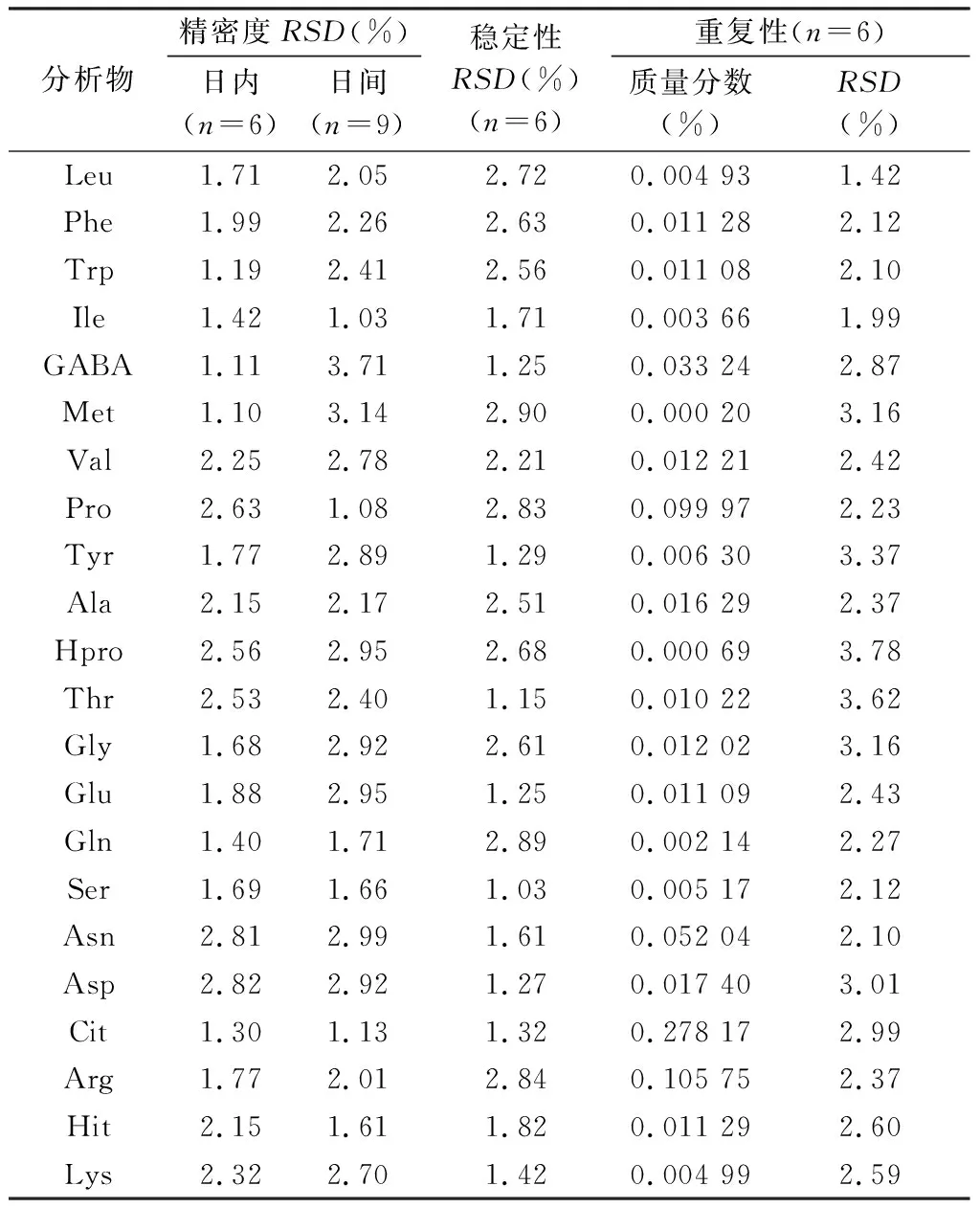
表3 22個氨基酸的精密度、重復性、穩定性結果
2.3.6 回收率試驗 精密稱取“2.3.5”項下已測知含量的栝樓桂枝顆粒6份,約0.25 g,按近似1∶1質量濃度精密加入22種對照品溶液,按“2.2.3”項下方法制備供試品溶液,按“2.1”項下條件進樣測定待測成分的量,計算回收率,結果見表4。
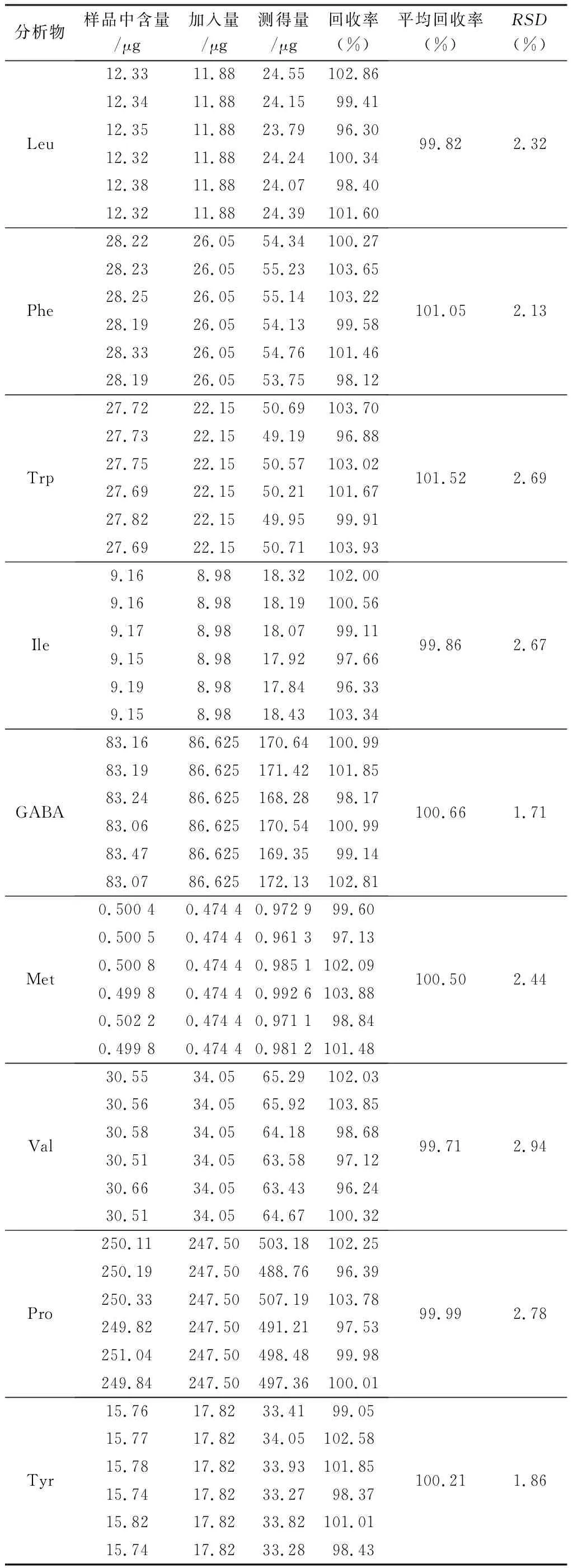
表4 栝樓桂枝顆粒中22種待測成分的回收率試驗結果(n=6)
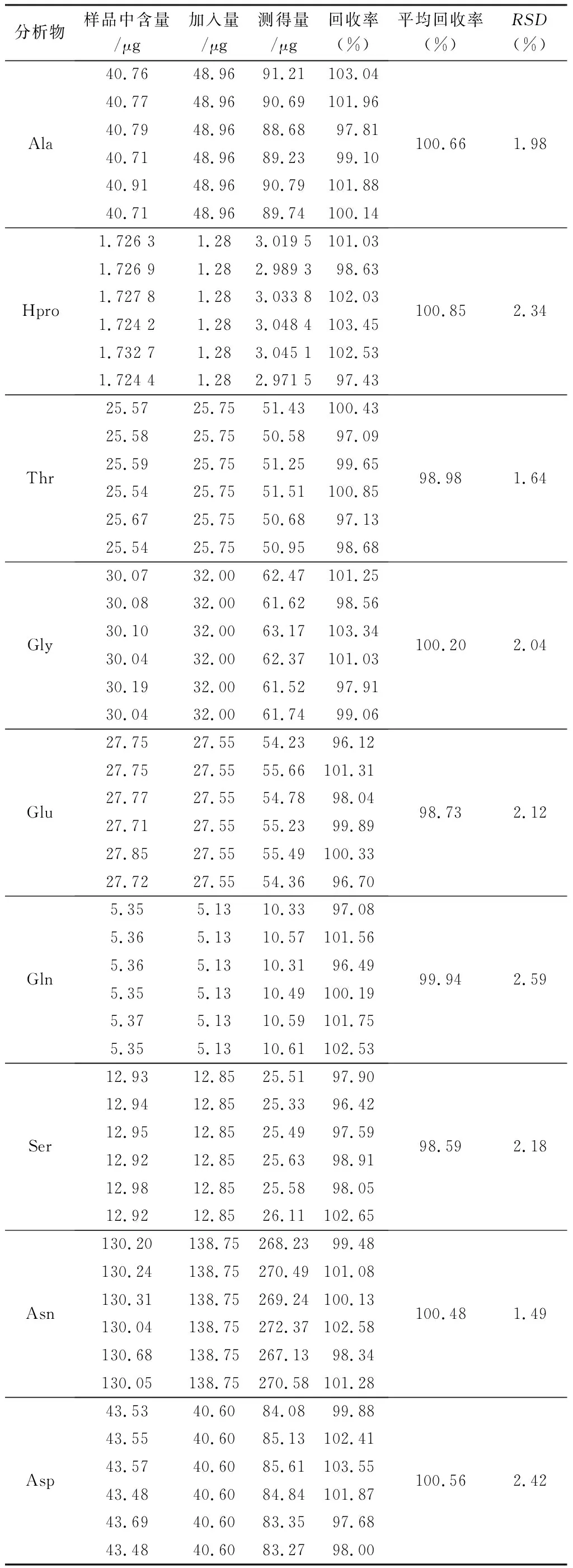
表4(續)
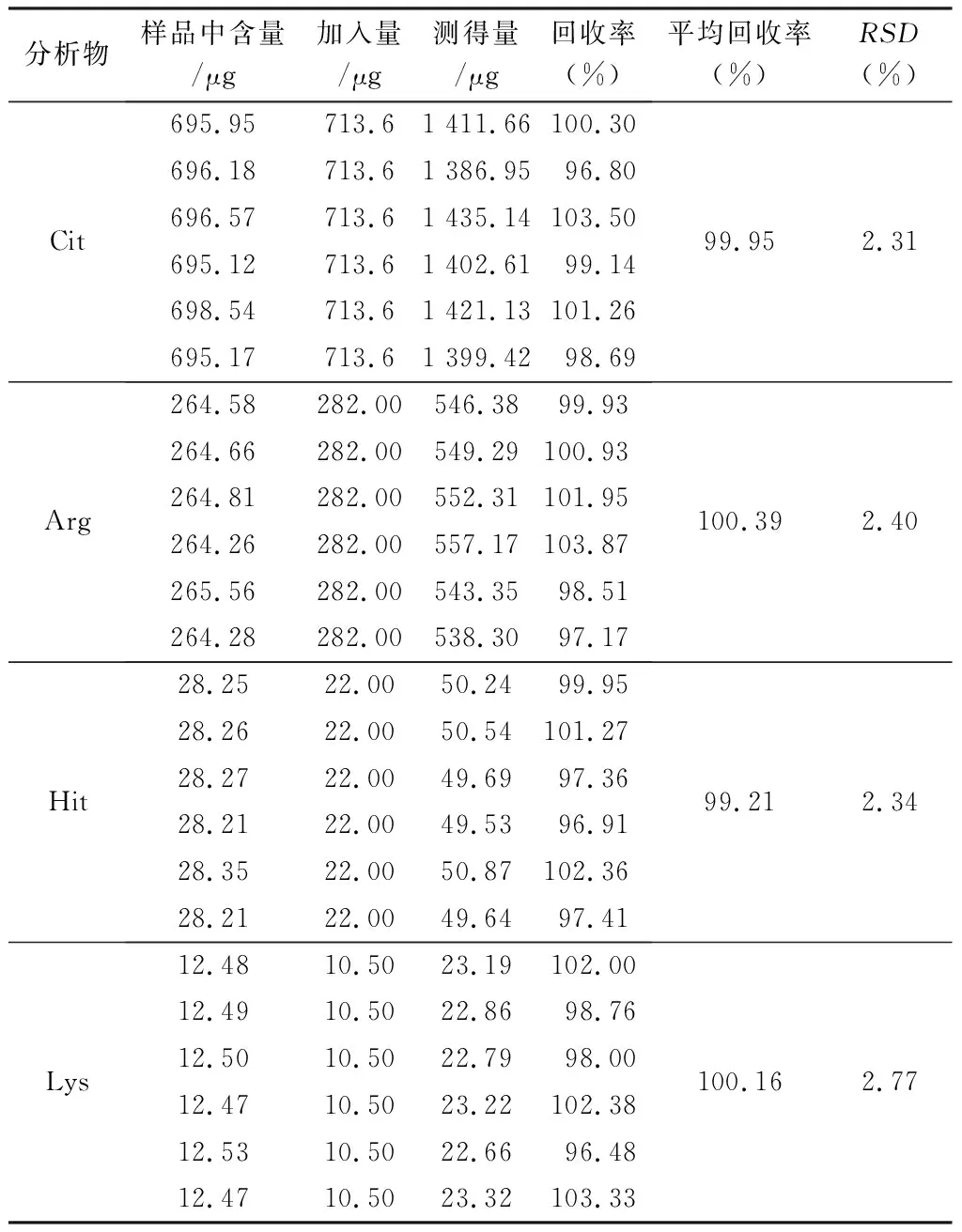
表4(續)
2.4 樣品含量測定 分別精密稱取0.5 g栝樓桂枝顆粒6批樣品粉末各3份,分別按“2.2.3”項下方法制備供試品溶液,按“2.1”項下條件進樣測定峰面積,根據內標法計算各批次栝樓桂枝顆粒中22種化合物的含量,結果見表5。

表5 6批栝樓桂枝顆粒各成分的含量(mg·g-1,n=3)
3 討論
本研究對22個氨基酸進行無衍生化的含量測定,首先色譜分離上,通過比較兩種類型的HILIC柱,包括BEH Amid酰胺HILIC柱(2.1 mm×100 mm,1.7 μm)和BEH 硅膠HILIC柱(2.1 mm×100 mm,1.7 μm),結果發現在相同的流動相中,前者對這些氨基酸類化合物的總體保留能力和分離度均優于后者;比較了流動相的不同添加劑,如甲酸銨、乙酸銨、甲酸、乙酸和氨水,結果顯示,在水中添加0.25%甲酸和2 mmol·L-1甲酸銨可得到的最佳峰形。其次在質譜檢測的優化上,發現大多數α-氨基酸均表現分子離子峰主要為[M+H]+峰,二級質譜上最佳子離子為結構中中性丟失一分子的甲酸,產生[M+H-HCOOH]+的子離子峰;而對于一些非α-氨基酸,如GABA,其子離子[M+H-NH3]+比[M+H-HCOOH]+豐度更高,最佳子離子為[M+H-NH3]+;另外含酰胺基團的氨基酸類化合物如Gln,能夠同時產生m/z130[M+H-NH3]+,m/z101[M+H-HCOOH]+和m/z84[M+H-HCOOH-NH3]+的子離子峰,其中[M+H-HCOOH-NH3]+的豐度最大,所以選其最佳定量子離子為[M+H-HCOOH-NH3]+,最終在色譜和質譜條件的優化下,在6 min內使22個測定的氨基酸成分得到準確的定量。
氨基酸已被證實是腦卒中的重要代謝物之一[16],興奮性毒性損傷是缺血性腦卒中的主要發病機制之一[17],腦缺血時神經抑制性氨基酸如GABA、瓜氨酸等具有神經保護作用[18-19],臨床研究發現缺血性腦卒中患者血清中組氨酸、甘氨酸、精氨酸、L-蛋氨酸、谷氨酰胺、L-瓜氨酸等發生改變[20],提示氨基酸類成分在腦卒中調控中可能扮演重要角色,通過本次含量測定發現栝樓桂枝顆粒中氨基酸含量較高的成分為瓜氨酸(Cit),含量范圍達到2.78~4.29 mg·g-1,此外γ-氨基丁酸(GABA)、脯氨酸(Pro)、天冬酰胺(Asn)、精氨酸(Arg)含量也較高,均達到0.3 mg·g-1以上,而亮氨酸(Leu)、異亮氨酸(Ile)、蛋氨酸(Met)、酪氨酸(Tyr)、羥脯氨酸(Hpro)、谷氨酰胺(Gln)、絲氨酸(Ser)、賴氨酸(Lys)含量較低,均低于0.1 mg·g-1。
本研究所建立的UHILIC-MS/MS法,可快速準確的測定栝樓桂枝顆粒中22個氨基酸含量,該方法相比于已有的氨基酸分析方法,經簡單的提取步驟即可制備樣品溶液,可有效避免氨基酸衍生化所帶來的弊端,方法可為快速測定中藥復方中氨基酸類成分提供方法技術,為揭示栝樓桂枝顆粒中氨基酸類成分的物質基礎奠定實驗依據。
