B36團簇組裝一維納米線的密度泛函研究
2022-03-04 13:23:18劉會霞陳文浩段海明
原子與分子物理學報 2022年3期
劉會霞, 陳文浩, 馬 潔, 段海明
(新疆大學 物理科學與技術學院, 烏魯木齊 830046)
1 引 言
硼(B)是元素周期表中碳的近鄰,單質硼塊體具有多種同素異形體[1]. 包含較少(40以下)原子數目的B團簇傾向于形成平面或者準平面的二維(2D)結構,在這些(準)二維結構中三角形框架為其典型結構特征[2,3]. 然而,隨著團簇尺寸的增加,純二維硼團簇中會出現(xiàn)多邊形(四、五、六邊形)空位[4-6]. 光電子能譜實驗和嚴格的理論計算均揭示出B36團簇為包含中心六方孔的六邊形“碗狀”準平面彎曲構型,具有高穩(wěn)定性和較高的(C6v)對稱性[7]. Liu等人通過密度泛函理論(DFT)研究,探討了六方孔出現(xiàn)在B36團簇中心位置的原因,并且表征了B36團簇在熱力學和動力學上也高度穩(wěn)定的[8]. 王來生教授團隊的研究表明,以B36團簇作為結構基元擴展形成包含六邊形空位的二維B單層結構也是可行的[7].
自從發(fā)現(xiàn)碳納米管[9]以來,納米線、納米棒等一維結構材料因其獨特的力學、熱學、光學、電子輸運等特性而備受矚目[10-18]. 傳統(tǒng)上制備納米線的實驗方法諸多,比如汽液固生長法、氧化物輔助生長法和碳熱反應法等[19]. 近年來,團簇組裝納米線方法越來越多地引起研究者的關注,以高穩(wěn)定團簇作為結構單元組成的一維納米結構材料具有獨特的性能和巨大的潛在應用價值[20,21]. 通常,單質納米線自身的帶隙過小,限制了其在納米電子器件方面的應用,對此,可以通過摻雜和化學修飾等方式改變原有納米線的電子結構[22],這對于提升納米線在納米器件領域的應用具有明顯的重要性和可行性[23,24].
盡管目前關于團簇組裝納米線的研究成果已有諸多報道,但是以B團簇作為結構單元組裝納米線的相關研究還很少見[25]. 據我們調研,至目前尚無B36團簇組裝納米線的相關研究工作發(fā)表. 考慮到實驗上B36團簇已經成功合成[7]、理論研究也表征了B36團簇具備高的熱穩(wěn)定性[8]和特殊的鍵合特性[26],本文即采用第一性原理計算方法系統(tǒng)研究了B36團簇組裝一維納米線體系的幾何結構與電子結構.
本文中,首先計算分析了孤立B36團簇的電子結構,而后研究了根據不同鏈接方式組裝B36團簇形成的兩類不同納米線,發(fā)現(xiàn)二者能量近簡并、但分別顯示出半金屬和小帶隙半導體特性,且二者均為熱力學穩(wěn)定體系. 此外,對兩種B36團簇組裝納米線進行了H原子吸附,發(fā)現(xiàn)吸附后的納米線均呈現(xiàn)出較大帶隙半導體特征.
2 計算方法
采用基于密度泛函理論的第一性原理計算方法. 具體計算使用VASP軟件包(Vienna Ab initio simulation package)軟件包. 采用投影綴加平面波(PAW)勢描述電子-離子相互作用,電子交換關聯(lián)勢選用廣義梯度近似(GGA)下的PBE形式[27-30],平面波截斷能設置為500 eV,幾何結構優(yōu)化中能量和力的收斂標準分別為10-6eV和0.01 eV/?. 布里淵區(qū)網格密度取1×1×5. 為表征體系的熱力學穩(wěn)定性,采用NVT系綜,在室溫300 K下,時間步長取為1 fs,模擬總時間長度為3.5 ps,使用第一性原理恒溫分子動力學方法計算體系的能量變化行為.
3 結果、分析和討論
3.1 B36團簇
圖1中給出了B36團簇的幾何結構示意圖及分波態(tài)密度圖. B36團簇具C6v對稱性,自內向外各六邊形環(huán)上獨立的B-B鍵長為1.662 ?(最內層)、1.670 ?(中間層),1.589 ?與1.673 ?(最外層). B36團簇并非為一完整平面構型,而是一個類似于“碗狀”彎曲的準平面結構. 計算所得B36團簇的幾何結構與之前Piazza等人的研究結果[7]是一致的. 分析圖1(c)可見,B36團簇具有1.07 eV的能隙,且靠近Feimi能的占據態(tài)主要由B原子的Pz軌道貢獻.

圖1 B36 團簇的幾何結構示意圖((a)為頂視圖、(b)為側視圖及分波態(tài)密度圖(c))Fig. 1 The structure diagrams, top view (a) and side view (b), and the partial density of states (c) of B36 cluster
3.2 B36團簇組裝納米線
將類“碗狀”B36團簇作為獨立結構單元組成一維納米線初始構型有兩種方式:“碗口”同向型(均朝下、如圖2(a)所示)或相鄰“碗口”反向型(一上一下、如圖2(b)所示),可稱為I型納米線及II型納米線. 兩種類型的初始納米線經優(yōu)化后結構均發(fā)生了較明顯的變化,不再是“碗狀”團簇的簡單連接,而是呈現(xiàn)出不同“波浪式”的納米線(如圖2(c)、(d)所示).
計算B36團簇組裝一維納米線平均結合能如下:
Eb=(nEB-Enano)/n
(1)
式中,EB為單個B原子的總能量,Enano為納米線的總能量,n為納米線所含B原子數. 計算所得I型納米線和II型納米線的平均結合能均為5.79 eV,表明這兩種類型的納米線具有相同的能量競爭性.
為了探討兩類不同納米線的熱力學穩(wěn)定性,采用第一性原理恒溫分子動力學方法計算了兩類納米線的能量演化行為. 基于NVT系綜,時間步長為1 fs,總模擬時間為3.5 ps,溫度取為300 K. 圖3給出兩類納米線的溫度-時間和能量-時間變化行為,可見,兩類納米線的能量各圍繞某一恒定值(~437.5 eV及~438 eV)做小的振蕩,均未出現(xiàn)能量的整體(向下)移動. 表明室溫下兩類納米線均為熱力學穩(wěn)定的,其動力學演化構型均可以視為相對初始穩(wěn)定結構的微小偏離.

圖2 B36 團簇組裝一維納米線的幾何結構示意圖. (a)、(b)為優(yōu)化前的初始結構,(c)、(d)為優(yōu)化后結構. (上為頂視圖、下為側視圖)Fig. 2 The structure diagrams of one-dimensional nanowires assembled by B36 clusters. (a) and (b) are the un-optimized initial structures, and (c) and (d) are the optimized final structures. (Top view shown above and side view at the bottom)
為表征B36團簇組裝一維納米線的電子結構,分別計算了兩類納米線的能帶結構和分波態(tài)密度,結果示于圖4中. 由圖4(a)可見I型納米線為一近零帶隙的半金屬,其費米能級附近電子態(tài)主要由B原子p軌道電子貢獻,在較深能級(距Feimi能約1eV之外)區(qū)域有B原子s軌道電子貢獻(見圖4(b)). 與I型納米線明顯不同,如圖4(c)所示,II型納米線的能帶圖呈現(xiàn)出直接帶隙半導體特征,具有約0.21 eV的較小帶隙. II型納米線Feimi能附件的電子態(tài)密度分布與I型納米線相類似,如圖4(d)所示,費米能級毗鄰處電子態(tài)主要由B原子p軌道貢獻,幾乎無B原子s軌道貢獻,而在較深能級區(qū)間可以看到B原子s軌道的明顯貢獻. 可見,以兩種不同組裝方式構造的兩類B36團簇組裝一維納米線具有不同的導電性能,分別為近零帶隙的半金屬和小帶隙半導體.
3.3 H原子吸附B36團簇組裝納米線
對于B36團簇組裝一維納米線吸附H原子體系,考慮將H原子吸附在I型及II型納米線“碗尖”位置B原子上,這與Yu等人研究α-硼烯納米線吸附H原子的情形是類似的[23]. 圖5給出了兩種類型的B36團簇組裝一維納米線吸附H原子體系的幾何結構示意圖,吸附H原子后納米線的幾何結構與未吸附時的結構相較變化甚微,依舊保持原有納米線的“波浪形”結構,可將其分別命名為I型吸H納米線和II型吸H納米線.
以如下公式計算I型及II型吸H納米線中單個H原子的吸附能:
Eads=(mEH+Enano(B)-Enano(B@H))/m
(2)
其中,EH指為單個H原子的總能量,Enano(B)指為原(未吸附H)納米線的總能量,Enano(B@H)指吸H納米線的總能量,m為(計算單元中)H原子數目.
計算所得I型及II型吸H納米線對H的吸附能均為2.97 eV,體現(xiàn)出二者對H的相同的相互作用.

圖3 B36 團簇組裝一維納米線的溫度及能量隨時間的變化((a)為I型納米線、(b)為II型納米線)Fig. 3 Variations of temperature and energy with time of one-dimensional nanowires assembled by B36 clusters for (a) the type-I nanowire and (b) the type-II nanowire
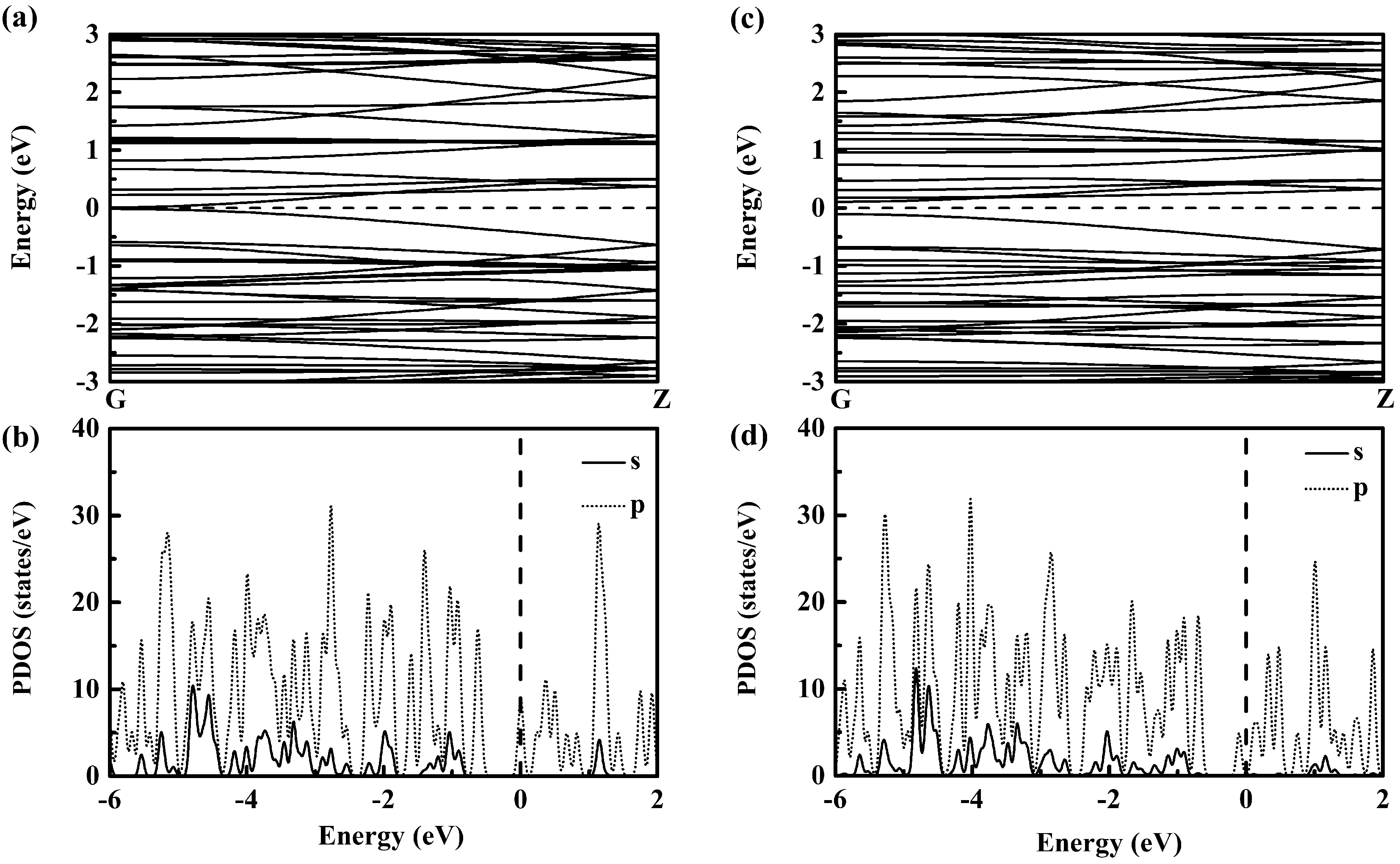
圖4 B36團簇組裝一維納米線的能帶及分波態(tài)密度((a)與(b)為I型納米線、(c)與(d)為II型納米線)Fig. 4 Energy band and partial density of states of one-dimensional nanowires assembled by B36 clusters for (a) and (b) the type-I nanowire and (c) and (d) the type-II nanowire

圖5 H吸附B36團簇組裝一維納米線的幾何結構示意圖. (a)、(b)為優(yōu)化前的初始結構,(c)、(d)為優(yōu)化后結構. (上為頂視圖、下為側視圖)Fig.5 The structure diagrams of H-adsorbed one-dimensional nanowires assembled by B36 clusters. (a) and (b) are the un-optimized initial structures, and (c) and (d) are the optimized final structures. (Top view shown above and side view at the bottom)
與未吸附H納米線情形類似,為探究吸H納米線的熱力學穩(wěn)定性,進行了同樣(溫度300 K、時間步長1 fs、總時長3.5 ps)的分子動力學模擬. 所得I型及II型吸H納米線的溫度及能量演化行為示于圖6中.
分析圖6可見,在運用恒溫分子動力學模擬的初始調溫階段(~500步之前)、伴隨著較大的溫度漲落兩類吸H納米線的能量變化也出現(xiàn)較大的波動,但在溫度趨于恒定后(~500步之后),兩類吸H納米線的能量漲落亦均做微小變化,能量均值基本恒定. 分析該恒溫過程中兩類納米線的動力學結構,發(fā)現(xiàn)其與納米線初始結構相比較并無明顯變化,這說明兩類吸H納米線在室溫下均是熱力學穩(wěn)定的.
H吸附通常會飽和吸附體上相鄰原子的懸掛鍵電子,導致體系電子的重新分布、影響及改變體系的電子結構. 圖7中給出了I型及II型吸H納米線的能帶與分波態(tài)密度. 原先為半金屬的未吸附H的I型納米線,在吸附H后轉變?yōu)橹苯訋栋雽w(能隙為0.43 eV,見圖7(a)所示). 分析該I型吸H納米線的分波態(tài)密度圖(圖7(b))可見,鄰近Feimi能的占據態(tài)(非占據態(tài))主要由B原子p軌道貢獻,B及H的s軌道電子貢獻很少. B、H的s電子主要分布于較深能級處(-3 eV以下),體現(xiàn)出被吸附H原子與吸附體之間較強的相互作用. 對于未吸附H的II型納米線,其自身為半導體(具有約0.21 eV的較小帶隙),在吸附H后還保持半導體特性,為直接帶隙半導體(能隙~0.42 eV,見圖7(c)所示). 該II型吸H納米線的分波態(tài)密度結構(圖7(d))與I型吸H納米線相類似:與Feimi能毗鄰的占據態(tài)及非占據態(tài)基本由來源于B原子的p軌道貢獻,B、H的s電子主體分布于較深能級處(-3eV以下),也體現(xiàn)出被吸附H原子與吸附體之間較強的相互作用.

圖6 H吸附B36 團簇組裝一維納米線的溫度及能量隨時間的變化((a)為I型吸H納米線、(b)為II型吸H納米線)Fig. 6 Variations of temperature and energy with time of H-adsorbed one-dimensional nanowires assembled by B36 clusters for (a) the type-I H-adsorbed nanowire and (b) the type-II H-adsorbed nanowire
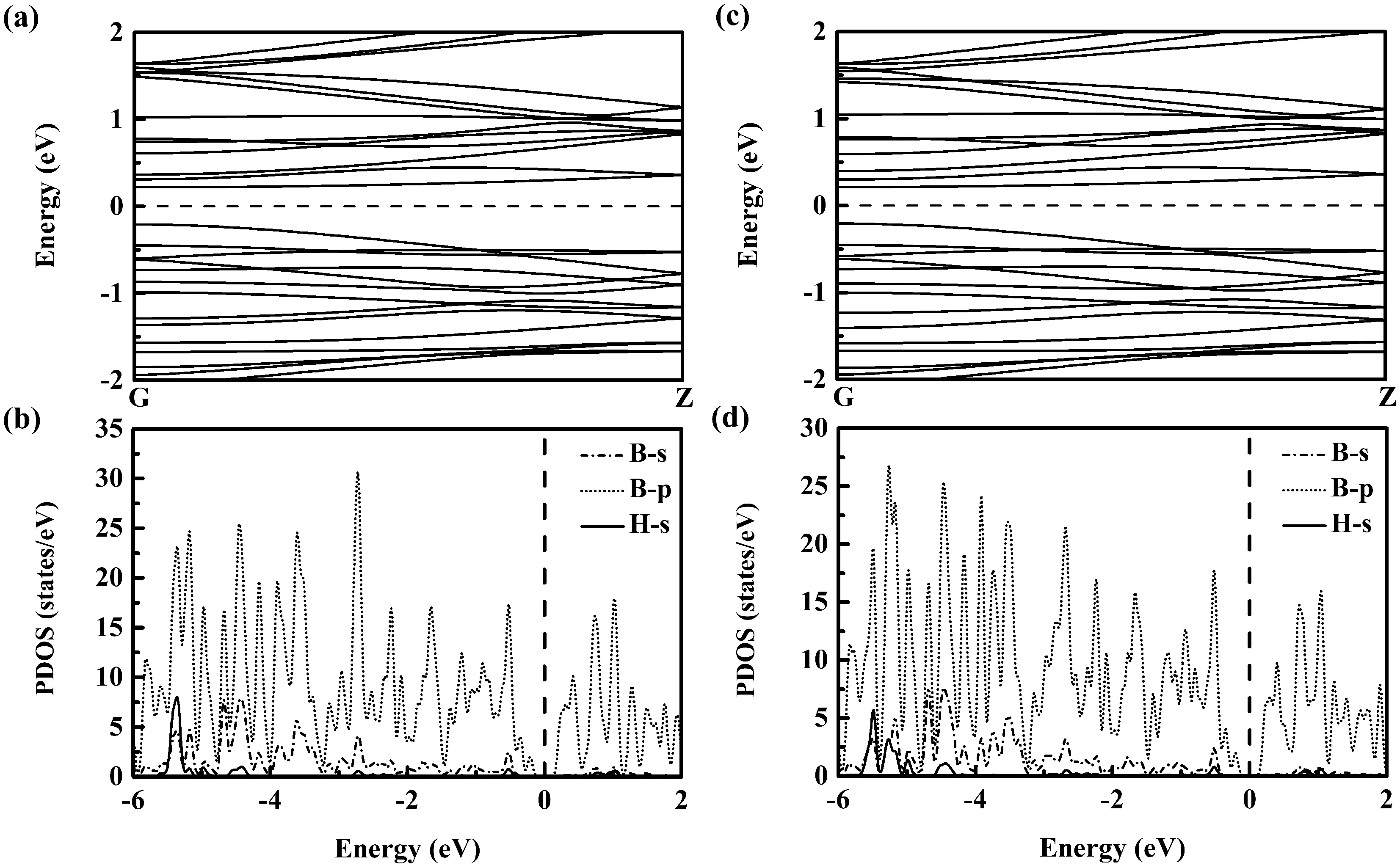
圖7 H吸附B36 團簇組裝一維納米線的能帶及分波態(tài)密度((a)與(b)為I型吸H納米線、(c)與(d)為II型吸H納米線)Fig. 7 Energy band and partial density of states of H-adsorbed one-dimensional nanowires assembled by B36 clusters for (a) and (b) the type-I H-adsorbed nanowire and (c) and (d) the type-II H-adsorbed nanowire
4 結 論
本文運用基于密度泛函理論的第一性原理計算方法系統(tǒng)研究了B36團簇組裝一維納米線及相應H吸附體系的幾何結構、電子結構及穩(wěn)定性. 以B36團簇最穩(wěn)定的“碗狀”結構為組裝單元,按照兩種不同對接方式得到兩類B36團簇組裝一維納米線. 二者具有相同的能量競爭優(yōu)勢,但二者電子結構顯著不同,分別為近零能隙的半金屬和小帶隙(~0.21 eV)的半導體. 室溫下的分子動力學模擬表明二者均為(室溫下)熱力學穩(wěn)定體系. 對于兩類H原子吸附B36團簇組裝一維納米線,二者均為直接帶隙半導體(帶隙分別為0.43 eV和0.42 eV),室溫下的分子動力學模擬表明二者亦均為熱力學穩(wěn)定體系. 可見,H原子吸附會改變體系的電子結構、增大體系的帶隙. 分析吸H納米線的分波態(tài)密度,發(fā)現(xiàn)H的s電子基本分布在較深能級處,體現(xiàn)出H原子與B納米線之間較強的相互作用.
猜你喜歡
哲學評論(2021年2期)2021-08-22 01:53:34
新世紀智能(數學備考)(2020年11期)2021-01-04 00:38:16
中華詩詞(2019年7期)2019-11-25 01:43:04
中國外匯(2019年17期)2019-11-16 09:31:14
模具制造(2019年3期)2019-06-06 02:10:54
影視與戲劇評論(2016年0期)2016-11-23 05:26:01
現(xiàn)代企業(yè)(2015年9期)2015-02-28 18:56:50
現(xiàn)代企業(yè)(2015年1期)2015-02-28 18:43:18
新高考·高一物理(2014年1期)2014-09-18 01:26:07
土木建筑工程信息技術(2013年2期)2013-10-17 03:14:12
