隔山消的指紋圖譜建立、化學(xué)模式識(shí)別分析及多指標(biāo)含量測(cè)定
2022-03-29 23:35:13孫佳勾健秦蘭劉亭潘潔韋宇劉春花
中國(guó)藥房 2022年6期
孫佳 勾健 秦蘭 劉亭 潘潔 韋宇 劉春花






中圖分類(lèi)號(hào) R284.1 文獻(xiàn)標(biāo)志碼 A 文章編號(hào) 1001-0408(2022)06-0673-07
DOI 10.6039/j.issn.1001-0408.2022.06.05
摘 要 目的 建立隔山消藥材的指紋圖譜并進(jìn)行化學(xué)模式識(shí)別分析,同時(shí)測(cè)定其中4種成分的含量。方法 采用高效液相色譜(HPLC)法。色譜柱為ACE UExcel C18,流動(dòng)相為乙腈-0.1%磷酸溶液(梯度洗脫),流速為1.2 mL/min,檢測(cè)波長(zhǎng)為210 nm,柱溫為40 ℃,進(jìn)樣量為10 μL。以青陽(yáng)參苷元為參照,采用《中藥色譜指紋圖譜相似度評(píng)價(jià)系統(tǒng)(2012版)》繪制16批隔山消藥材的HPLC指紋圖譜并進(jìn)行相似度評(píng)價(jià),確定共有峰。采用SPSS 26.0軟件和SIMCA 14.0軟件進(jìn)行聚類(lèi)分析、主成分分析和正交偏最小二乘-判別分析,以變量重要性投影(VIP)值大于1為標(biāo)準(zhǔn)篩選影響隔山消藥材質(zhì)量的差異性成分;采用相同的HPLC法測(cè)定丁香酸、去酰基蘿藦苷元、白首烏二苯酮和青陽(yáng)參苷元的含量。結(jié)果 16批隔山消藥材中共有29個(gè)共有峰,相似度為0.723~0.998;共指認(rèn)了4個(gè)共有峰,分別為丁香酸(7號(hào)峰)、去酰基蘿藦苷元(9號(hào)峰)、白首烏二苯酮(13號(hào)峰)、青陽(yáng)參苷元(15號(hào)峰)。聚類(lèi)分析結(jié)果顯示,16批隔山消藥材可聚為3類(lèi),其中S1為一類(lèi),S3為一類(lèi),S2、S4~S16為一類(lèi)。主成分分析結(jié)果顯示,5個(gè)主成分的累計(jì)方差貢獻(xiàn)率為88.706%,分類(lèi)結(jié)果與聚類(lèi)分析結(jié)果一致。正交偏最小二乘-判別分析結(jié)果顯示,VIP值大于1的共有峰由大到小依次為20號(hào)峰、10號(hào)峰、25號(hào)峰、12號(hào)峰、15號(hào)峰(青陽(yáng)參苷元)、21號(hào)峰、14號(hào)峰、16號(hào)峰、26號(hào)峰、22號(hào)峰、17號(hào)峰。丁香酸、去酰基蘿藦苷元、白首烏二苯酮和青陽(yáng)參苷元檢測(cè)質(zhì)量濃度的線性范圍分別為0.715 3~45.778 0、2.379 4~152.281 0、0.642 0~41.085 0、14.541 6~930.662 0 μg/mL(R 2均大于0.999);定量限分別為0.357 7、0.475 9、0.642 0、2.423 6 μg/mL,檢測(cè)限分別為0.146 0、0.164 1、0.248 8、0.833 3 μg/mL;精密度、重復(fù)性、穩(wěn)定性(24 h)試驗(yàn)的RSD均小于3%;平均加樣回收率分別為99.11%(RSD=2.00%,n=9)、98.54%(RSD=2.21%,n=9)、96.33%(RSD=2.54%,n=9)、95.96%(RSD=2.93%,n=9);含量分別為17.12~147.80、95.23~583.10(S8低于定量限)、16.91~210.88、211.68~3 587.15(S1低于定量限)μg/g。結(jié)論 所建HPLC指紋圖譜和含量測(cè)定方法穩(wěn)定、可靠、準(zhǔn)確度高且重復(fù)性好,結(jié)合化學(xué)模式識(shí)別分析可用于隔山消藥材的質(zhì)量控制。
關(guān)鍵詞 隔山消;高效液相色譜法;指紋圖譜;化學(xué)模式識(shí)別分析;含量測(cè)定
Establishment of fingerprint, analysis of chemical pattern recognition and multi-index content determination of Cynanchum auriculatum
SUN Jia1,GOU Jian2,QIN Lan2,LIU Ting1,PAN Jie3,WEI Yu2,LIU Chunhua3(1. Provincial Key Lab of Pharmaceutics in Guizhou Province/State Key Lab of Functions and Applications of Medicinal Plants, Guizhou Medical University, Guiyang 550004, China; 2. College of Pharmacy, Guizhou Medical University, Guiyang 550004, China; 3. Engineering Research Center of the Ministry of Education for the Development and Application of Ethnic Medicine and Traditional Chinese Medicine, Guizhou Medical University, Guiyang 550004, China)
ABSTRACT? ?OBJECTIVE To establish the fingerprint of Cynanchum auriculatum, to conduct its chemical pattern recognition analysis, and to determine the contents of four components at the same time. METHODS High performance liquid chromatography (HPLC) method was adopted. The determination was performed on ACE UExcel C18 column with mobile phase consisted of acetonitrile-0.1% phosphoric acid solution (gradient elution) at the flow rate of 1.2 mL/min. The determination wavelength was set at 210 nm, and the column temperature was 40 ℃. The sample size was 10 μL. Taking qingyangshengenin as the reference, HPLC fingerprints of 16 batches of C. auriculatum medicinal materials were drawn and similarity was evaluated by using the Similarity Evaluation of Chromatographic Fingerprints of Traditional Chinese Medicine (2012 edition), and the common peaks were determined. SPSS 26.0 software and SIMCA 14.0 software were used for cluster analysis, principal component analysis and orthogonal partial least squares- discriminant analysis. The differential components affecting the quality of C. auriculatum were screened by taking the value of variable importance in projection (VIP) greater than 1 as the standard; same HPLC method was used to determine the contents of syringic acid, acyl asclepiadelenin, baishouwubenzophenone and qingyangshengenin. RESULTS There were 29 common peaks in 16 batches of C. auriculatum, with a similarity of 0.723-0.998. Four common peaks were identified, namely syringic acid (peak 7), acyl asclepiadoidin (peak 9), baishouwubenzophenone (peak 13) and qingyangshengenin (peak 15). The results of cluster analysis showed that 16 batches of C. auriculatum could be clustered into three categories, among which S1 were grouped into one category, S3 were grouped into one category, S2, and S4-S16 were grouped into one category. The results of principal component analysis showed that the cumulative variance contribution rate of the five principal components was 88.706%, and the classification results were consistent with the results of cluster analysis. The results of orthogonal partial least squares-discriminant analysis showed that the common peaks (from large to small) with VIP value greater than 1 were peak 20, peak 10, peak 25, peak 12, peak 15 (qingyangshengenin), peak 21, peak 14, peak 16, peak 26, peak 22 and peak 17. The linear ranges of syringic acid, acyl asclepterin, baishouwubenzophenone and qingyangshengenin were 0.715 3-45.778 0, 2.379 4-152.281 0, 0.642 0- 41.085 0, 14.541 6- 930.662 0 μg/mL respectively (all R 2>0.999). The quantitative limits were 0.357 7, 0.475 9, 0.642 0 and 2.423 6 μg/mL; the detective limits were 0.146 0, 0.164 1, 0.248 8 and 0.833 3 μg/mL, respectively. RSDs of precision, repeatability and stability (24 h) tests were less than 3%; the average recoveries were 99.11% (RSD=2.00%, n=9), 98.54% (RSD=2.21%, n=9), 96.33% (RSD=2.54%, n=9) and 95.96% (RSD=2.93%, n=9); the contents were 17.12-147.80, 95.23-583.10 (S8 below the quantitative limit), 16.91-210.88 and 211.68-3 587.15 (S1 below the quantitative limit) μg/g, respectively. CONCLUSIONS Established HPLC fingerprint and the method of content determination are stable, reliable, accurate and reproducible. Combined with analysis of chemical pattern recognition, it can be used for the quality control of C. auriculatum.
KEYWORDS? ?Cynanchum auriculatum; high performance liquid chromatography; fingerprint; analysis of chemical pattern recognition; content determination
隔山消又名飛來(lái)鶴、過(guò)山消、隔山撬等,為蘿藦科植物耳葉牛皮消Cynanchum auriculatum Royle ex Wight的干燥塊根,味甘、苦,性微溫,主要分布于我國(guó)云南、貴州、四川等地的少數(shù)民族聚居區(qū)[1-2]。研究發(fā)現(xiàn),隔山消含有苯乙酮類(lèi)、甾體苷類(lèi)、有機(jī)酸類(lèi)、香豆素類(lèi)、木脂素類(lèi)等化學(xué)成分[1,3],具有補(bǔ)肝腎、強(qiáng)筋骨、益精血、烏須發(fā)和消積止痛等功效,民間多用于治療痢疾、食積飽脹、胃氣痛等胃腸道疾病,臨床用于治療虛損勞傷、腹痛腹脹、急慢性脅痛、腹水等癥[4-5]。隔山消現(xiàn)收載于《貴州省中藥材、民族藥材質(zhì)量標(biāo)準(zhǔn)》《中華本草·苗藥卷》《貴陽(yáng)民間藥草》等標(biāo)準(zhǔn)/書(shū)籍中[6-8],但上述標(biāo)準(zhǔn)/書(shū)籍僅對(duì)隔山消藥材的性狀、性味、鑒別等方面進(jìn)行了描述。本課題組前期研究發(fā)現(xiàn),隔山消提取物具有促進(jìn)胃腸道蠕動(dòng)的作用,其主要活性成分為白首烏二苯酮、去酰基蘿藦苷元、青陽(yáng)參苷元和丁香酸等[9]。由于中藥所含化學(xué)成分較為復(fù)雜,且質(zhì)量易受生產(chǎn)地域、采摘時(shí)間、炮制方式等因素的影響,從而導(dǎo)致藥材質(zhì)量參差不齊,加之目前關(guān)于隔山消藥材的研究多集中在化學(xué)成分及藥理作用等方面[9-11],有關(guān)含量測(cè)定的研究有限且僅以單一成分或單一種類(lèi)化合物作為指標(biāo)[12-13],因此,該藥材缺乏更全面、專(zhuān)屬性更強(qiáng)的質(zhì)量評(píng)價(jià)方法。
中藥指紋圖譜具有整體性、穩(wěn)定性的優(yōu)點(diǎn),可完整、系統(tǒng)地表征藥材樣品中主要化學(xué)成分的相似性[14-15]。化學(xué)模式識(shí)別方法近年來(lái)被廣泛應(yīng)用于中藥材及中藥制劑的質(zhì)量評(píng)價(jià)中,可對(duì)多種現(xiàn)代儀器分析所得數(shù)據(jù)進(jìn)行客觀分析,并將其量化[14,16-17]。基于此,本研究建立了隔山消藥材的高效液相色譜(high performance liquid chromatography,HPLC)指紋圖譜,同時(shí)結(jié)合化學(xué)模式識(shí)別進(jìn)行分析,并采用相同HPLC法測(cè)定了藥材中白首烏二苯酮、去酰基蘿藦苷元、青陽(yáng)參苷元和丁香酸的含量,旨在為隔山消藥材的質(zhì)量控制提供參考。
1 材料
1.1 主要儀器
本研究所用主要儀器有U3000型HPLC儀及配備的四元泵、自動(dòng)進(jìn)樣器、柱溫箱、脫氣器、光電二極管陣列檢測(cè)器(美國(guó)Thermo Fisher Scientific公司),Allegra X-30R Centrifuge型高速離心機(jī)(美國(guó)Beckman Coulter公司),DK-98-Ⅱ型電熱恒溫水浴鍋(天津市泰斯特儀器有限公司),KQ-300DE型數(shù)控超聲波清洗器(昆山市超聲儀器有限公司),EL204型電子天平[梅特勒-托利多儀器(上海)有限公司],WP-UP-Ⅱ-20型超純水機(jī)(四川沃特爾水處理設(shè)備有限公司)等。
1.2 主要藥品與試劑
丁香酸對(duì)照品(批號(hào)PS010564,純度≥98%)購(gòu)自成都普思生物科技股份有限公司;去酰基蘿藦苷元對(duì)照品(批號(hào)CS-200723,純度≥99.66%)購(gòu)自北京世紀(jì)奧科生物技術(shù)有限公司;白首烏二苯酮對(duì)照品(純度≥98%)由貴州醫(yī)科大學(xué)貴州省藥物制劑重點(diǎn)實(shí)驗(yàn)室自制;青陽(yáng)參苷元對(duì)照品(批號(hào)AF20062803,純度≥98%)購(gòu)自成都埃法生物科技有限公司;甲醇、乙醇、乙腈、磷酸均為色譜純,其余試劑均為分析純,水為超純水。
16批隔山消藥材(編號(hào)S1~S16)中,S11~S14批樣品購(gòu)自貴州飛云嶺藥業(yè)股份有限公司,其余批次藥材購(gòu)自貴州、四川及云南等地的藥材市場(chǎng)。上述藥材購(gòu)于2018年3月-2020年8月,經(jīng)貴州醫(yī)科大學(xué)藥學(xué)院生藥學(xué)教研室劉春花副教授鑒定為蘿藦科植物隔山消C. auriculatum Royle ex Wight的干燥塊根。16批隔山消藥材的來(lái)源信息見(jiàn)表1。
2 方法與結(jié)果
2.1 隔山消藥材的HPLC指紋圖譜建立
2.1.1 色譜條件 以ACE UExcel C18(250 mm×4.6 mm,5 μm)為色譜柱,以乙腈(A)-0.1%磷酸溶液(B)為流動(dòng)相進(jìn)行梯度洗脫(0~5 min,10%A→15%A;5~25 min,15%A→30%A;25~35 min,30%A→42%A;35~55 min,42%A→90%A;55~60 min,90%A;60~70 min,90%A→10%A);流速為1.2 mL/min;檢測(cè)波長(zhǎng)為210 nm;柱溫為40 ℃;進(jìn)樣量為10 μL。
2.1.2 混合對(duì)照品溶液的制備 取丁香酸、去酰基蘿藦苷元、白首烏二苯酮、青陽(yáng)參苷元對(duì)照品適量,精密稱(chēng)定,加甲醇溶解并定容,制成上述4種成分質(zhì)量濃度分別為45.778 0、152.281 0、41.085 0、930.662 0 μg/mL的混合對(duì)照品溶液,于-20 ℃儲(chǔ)存,備用。
2.1.3 供試品溶液的制備 取隔山消藥材粉末(過(guò)60目篩,下同),約1.0 g,置于具塞錐形瓶中,加入0.1 mol/L鹽酸6 mL、30%乙醇50 mL,加熱回流提取4 h,濾過(guò),用30%乙醇20 mL洗滌濾渣,合并濾液和洗液,水浴揮至近干,殘?jiān)蛹状既芙獠⒍ㄈ葜? mL,以20 000 r/min離心10 min,取上清液,即得供試品溶液。
2.1.4 精密度試驗(yàn) 取“2.1.3”項(xiàng)下供試品溶液(編號(hào)S7),按“2.1.1”項(xiàng)下色譜條件連續(xù)進(jìn)樣測(cè)定6次,以青陽(yáng)參苷元為參照,計(jì)算各共有峰的相對(duì)保留時(shí)間和相對(duì)峰面積。結(jié)果顯示,29個(gè)共有峰相對(duì)保留時(shí)間的RSD為0.02%~1.53%,相對(duì)峰面積的RSD為0.56%~2.50%(n=6),表明方法精密度良好。
2.1.5 重復(fù)性試驗(yàn) 取隔山消藥材(編號(hào)S7)粉末1.0 g,共6份,精密稱(chēng)定,按“2.1.3”項(xiàng)下方法制備供試品溶液,再按“2.1.1”項(xiàng)下色譜條件進(jìn)樣測(cè)定,以青陽(yáng)參苷元為參照,計(jì)算各共有峰的相對(duì)保留時(shí)間和相對(duì)峰面積。結(jié)果顯示,29個(gè)共有峰相對(duì)保留時(shí)間的RSD為0.15%~1.41%,相對(duì)峰面積的RSD為1.20%~2.86%(n=6),表明方法重復(fù)性良好。
2.1.6 穩(wěn)定性試驗(yàn) 取“2.1.3”項(xiàng)下供試品溶液(編號(hào)S7),分別于室溫下放置0、2、4、8、12、24 h時(shí)按“2.1.1”項(xiàng)下色譜條件進(jìn)樣測(cè)定,以青陽(yáng)參苷元為參照,計(jì)算各共有峰的相對(duì)保留時(shí)間和相對(duì)峰面積。結(jié)果顯示,29個(gè)共有峰相對(duì)保留時(shí)間的RSD為0.07%~2.53%,相對(duì)峰面積的RSD為1.41~2.81%(n=6),表明供試品溶液于室溫下放置24 h內(nèi)穩(wěn)定性良好。
2.1.7 指紋圖譜的建立 取16批隔山消藥材,按“2.1.3”項(xiàng)下方法制備供試品溶液,再按“2.1.1”項(xiàng)下色譜條件進(jìn)樣測(cè)定,采用《中藥色譜指紋圖譜相似度評(píng)價(jià)系統(tǒng)(2012版)》對(duì)所得隔山消的色譜圖進(jìn)行分析,以各成分色譜響應(yīng)均較強(qiáng)的S2樣品為參照?qǐng)D譜,采用中位數(shù)法生成對(duì)照指紋圖譜,設(shè)定時(shí)間窗寬度為0.1 s,經(jīng)多點(diǎn)校正和mark峰匹配生成隔山消指紋圖譜的共有模式并建立16批隔山消藥材的疊加指紋圖譜。結(jié)果顯示,16批隔山消藥材共有29個(gè)共有峰。16批隔山消藥材的HPLC疊加指紋圖譜見(jiàn)圖1,對(duì)照指紋圖譜見(jiàn)圖2。
2.1.8 共有峰指認(rèn) 取“2.1.2”項(xiàng)下混合對(duì)照品溶液,按“2.1.1”項(xiàng)下色譜條件進(jìn)樣測(cè)定,記錄色譜圖(圖3A)。將16批隔山消藥材的HPLC疊加指紋圖譜與上述混合對(duì)照品溶液色譜圖進(jìn)行對(duì)比,共指認(rèn)了其中4個(gè)色譜峰,分別為丁香酸(7號(hào)峰)、去酰基蘿藦苷元(9號(hào)峰)、白首烏二苯酮(13號(hào)峰)、青陽(yáng)參苷元(15號(hào)峰)。由于青陽(yáng)參苷元(15號(hào)峰)的分離度較好、峰面積較穩(wěn)定且保留時(shí)間適中,故以其為參照。
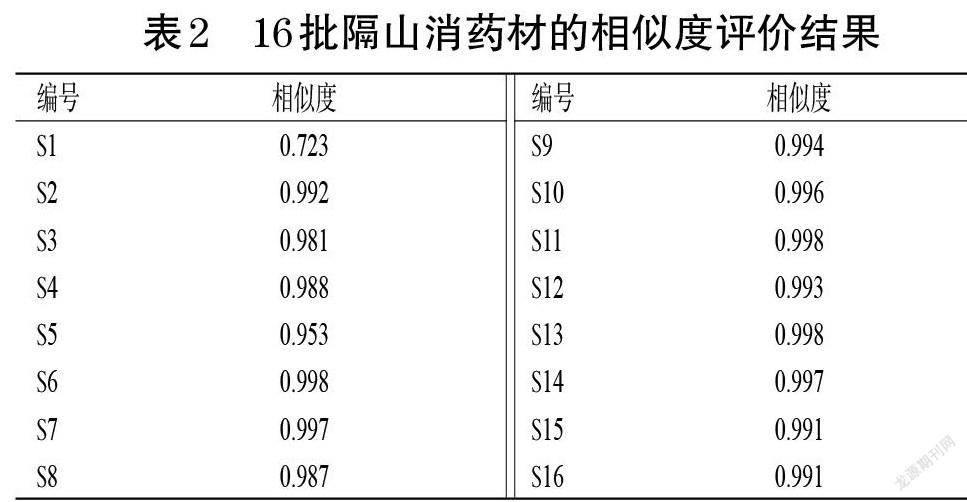
2.1.9 相似度評(píng)價(jià) 以對(duì)照指紋圖譜為參照,采用《中藥色譜指紋圖譜相似度評(píng)價(jià)系統(tǒng)(2012版)》對(duì)16批隔山消藥材的指紋圖譜進(jìn)行相似度評(píng)價(jià)。結(jié)果顯示,16批隔山消藥材的相似度為0.723~0.998,其中僅S1的相似度為0.723,其余均大于0.950,表明除S1外,其余15批隔山消藥材的質(zhì)量穩(wěn)定、化學(xué)成分差異較小。結(jié)果見(jiàn)表2。
2.2 聚類(lèi)分析
以16批隔山消藥材指紋圖譜的共有峰峰面積為變量,使用SPSS 26.0軟件,采用瓦爾德連接方法(即離差平方和法),以平方歐氏距離為測(cè)度進(jìn)行聚類(lèi)分析。結(jié)果顯示,當(dāng)距離為10時(shí),16批隔山消藥材可聚為3類(lèi),其中S1為一類(lèi),S3為一類(lèi),S2、S4~S16為一類(lèi),表明S1、S3與其余14批隔山消藥材質(zhì)量存在差異。結(jié)果見(jiàn)圖4。
2.3 主成分分析
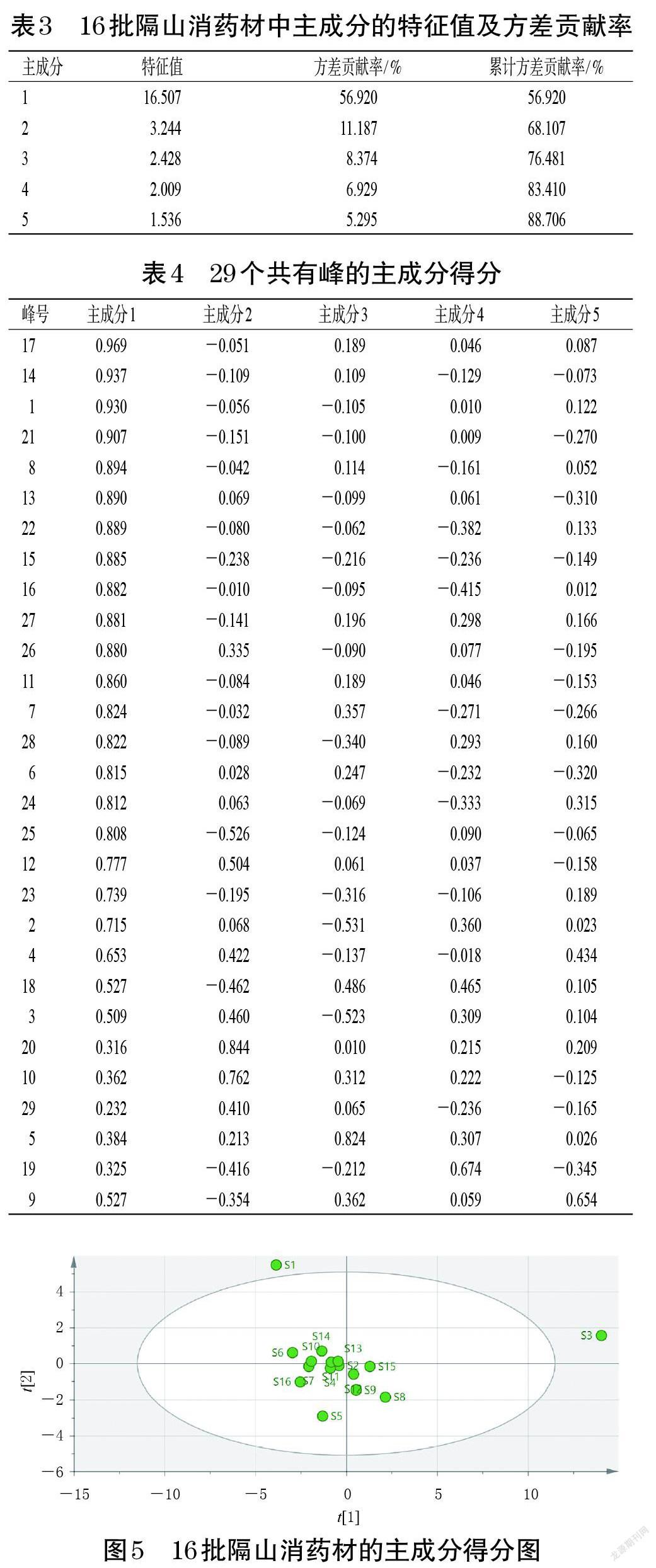
以16批隔山消藥材指紋圖譜的共有峰峰面積為變量,使用SPSS 26.0軟件進(jìn)行主成分分析。結(jié)果顯示,共提取5個(gè)主成分,其中主成分1的特征值最大,為16.507,方差貢獻(xiàn)率為59.920%,主要反映1~4、6~8、11~18、21~28號(hào)峰的信息。主成分2~5的特征值分別為3.244、2.428、2.009、1.536,方差貢獻(xiàn)率分別為11.187%、8.374%、6.929%、5.295%,其中主成分2反映10、20、29號(hào)峰的信息,主成分3反映5號(hào)峰的信息,主成分4反映19號(hào)峰的信息,主成分5反映9號(hào)峰的信息。5個(gè)主成分的累計(jì)方差貢獻(xiàn)率為88.706%,表明提取的這5個(gè)主成分可以反映樣品數(shù)據(jù)中的絕大部分信息,能代表16批隔山消藥材樣品中29個(gè)共有峰峰面積,提示隔山消藥材質(zhì)量受多個(gè)成分的影響。結(jié)果見(jiàn)表3、表4 。
以16批隔山消藥材指紋圖譜的共有峰峰面積為變量,采用SIMCA 14.0軟件繪制主成分得分圖,詳見(jiàn)圖5。由圖5可知,16批隔山消藥材可分為3類(lèi),S1樣品位于得分圖的左上方,為一類(lèi);S3樣品位于得分圖的最右側(cè),為一類(lèi);S2、S4~S16樣品位于得分圖的中部,為一類(lèi)。
2.4 正交偏最小二乘-判別分析
以16批隔山消藥材指紋圖譜的共有峰峰面積為變量,使用SIMCA 14.0軟件進(jìn)行正交偏最小二乘-判別分析。結(jié)果顯示,累計(jì)解釋能力參數(shù)R 2X、R 2Y分別為0.867、0.975,累計(jì)模型預(yù)測(cè)能力參數(shù)Q 2為0.552,均大于0.5,表明該模型穩(wěn)定、可靠[18]。16批隔山消藥材被分為3類(lèi),其中S1為一類(lèi),S3為一類(lèi),S2、S4~S16為一類(lèi)。結(jié)果見(jiàn)圖6。
變量重要性投影(variable importance in projection,VIP)值是篩選差異性成分的重要指標(biāo),VIP值越高,成分對(duì)組間差異的影響越大,故本研究以VIP值>1為標(biāo)準(zhǔn)篩選影響隔山消藥材質(zhì)量的差異性成分[14,18]。結(jié)果顯示,VIP值>1的共有峰由大到小依次為20號(hào)峰、10號(hào)峰、25號(hào)峰、12號(hào)峰、15號(hào)峰(青陽(yáng)參苷元)、21號(hào)峰、14號(hào)峰、16號(hào)峰、26號(hào)峰、22號(hào)峰、17號(hào)峰。結(jié)果見(jiàn)圖7。
2.5 隔山消藥材中4個(gè)有效成分的含量測(cè)定
2.5.1 色譜條件 同“2.1.1”項(xiàng)。
2.5.2 混合對(duì)照品溶液的制備 同“2.1.2”項(xiàng)。
2.5.3 供試品溶液的制備 同“2.1.3”項(xiàng)。
2.5.4 系統(tǒng)適用性試驗(yàn) 取上述混合對(duì)照品溶液、供試品溶液、空白對(duì)照溶液(按“2.5.3”項(xiàng)下方法制備不含隔山消藥材的空白對(duì)照溶液),按“2.5.1”項(xiàng)下色譜條件進(jìn)樣測(cè)定,記錄色譜圖,詳見(jiàn)圖3。由圖3可知,在供試品溶液中,丁香酸等4個(gè)成分的保留時(shí)間與對(duì)照品溶液的保留時(shí)間一致,分離度均大于1.5,理論板數(shù)均不低于7 000,空白對(duì)照溶液不影響待測(cè)成分的含量測(cè)定。
2.5.5 線性關(guān)系考察 取“2.5.2”項(xiàng)下混合對(duì)照品溶液適量,用50%甲醇逐級(jí)稀釋?zhuān)频枚∠闼豳|(zhì)量濃度分別為22.889 0、11.444 5、5.722 2、2.861 1、1.430 6、0.715 3 μg/mL,去酰基蘿藦苷元質(zhì)量濃度分別為76.140 5、38.070 2、19.035 1、9.517 6、4.758 8、2.379 4 μg/mL,白首烏二苯酮質(zhì)量濃度分別為20.542 5、10.271 3、5.135 6、2.567 8、1.283 9、0.642 0 μg/mL,青陽(yáng)參苷元質(zhì)量濃度分別為465.331 0、232.665 5、116.332 8、58.166 4、29.083 2、14.541 6 μg/mL的系列線性工作溶液。精密吸取上述系列線性工作溶液和“2.5.2”項(xiàng)下混合對(duì)照品溶液,按“2.5.1”項(xiàng)下色譜條件進(jìn)樣測(cè)定,記錄色譜圖。以各待測(cè)成分質(zhì)量濃度(x)為橫坐標(biāo)、峰面積(y)為縱坐標(biāo)進(jìn)行線性回歸,結(jié)果見(jiàn)表5。
2.5.6 定量限與檢測(cè)限考察 取“2.5.2”項(xiàng)下混合對(duì)照品溶液,用50%甲醇倍比稀釋?zhuān)础?.5.1”項(xiàng)下色譜條件進(jìn)樣測(cè)定,記錄峰面積。以信噪比分別為10 ∶ 1、3 ∶ 1計(jì)算定量限和檢測(cè)限,結(jié)果見(jiàn)表4。
2.5.7 精密度試驗(yàn) 取“2.5.3”項(xiàng)下供試品溶液(編號(hào)S7),按“2.5.1”項(xiàng)下色譜條件連續(xù)進(jìn)樣測(cè)定6次,記錄峰面積。結(jié)果顯示,丁香酸、去酰基蘿藦苷元、白首烏二苯酮、青陽(yáng)參苷元峰面積的RSD分別為1.00%、2.20%、1.80%、0.54%(n=6),表明方法精密度良好。
2.5.8 重復(fù)性試驗(yàn) 取隔山消藥材(編號(hào)S7)粉末1.0 g,共6份,精密稱(chēng)定,分別按“2.5.3”項(xiàng)下方法制備供試品溶液,再按“2.5.1”項(xiàng)下色譜條件進(jìn)樣測(cè)定,記錄峰面積并按外標(biāo)法計(jì)算樣品含量。結(jié)果顯示,丁香酸、去酰基蘿藦苷元、白首烏二苯酮、青陽(yáng)參苷元含量的RSD分別為0.67%、2.00%、2.80%、0.58%(n=6),表明方法重復(fù)性良好。
2.5.9 穩(wěn)定性試驗(yàn) 取“2.5.3”項(xiàng)下供試品溶液(編號(hào)S7),分別于室溫下放置0、2、4、8、12、24 h時(shí)按“2.5.1”項(xiàng)下色譜條件進(jìn)樣測(cè)定,記錄峰面積。結(jié)果顯示,丁香酸、去酰基蘿藦苷元、白首烏二苯酮、青陽(yáng)參苷元峰面積的RSD分別為1.40%、1.60%、2.40%、0.98%(n=6),表明供試品溶液于室溫下放置24 h內(nèi)穩(wěn)定性良好。
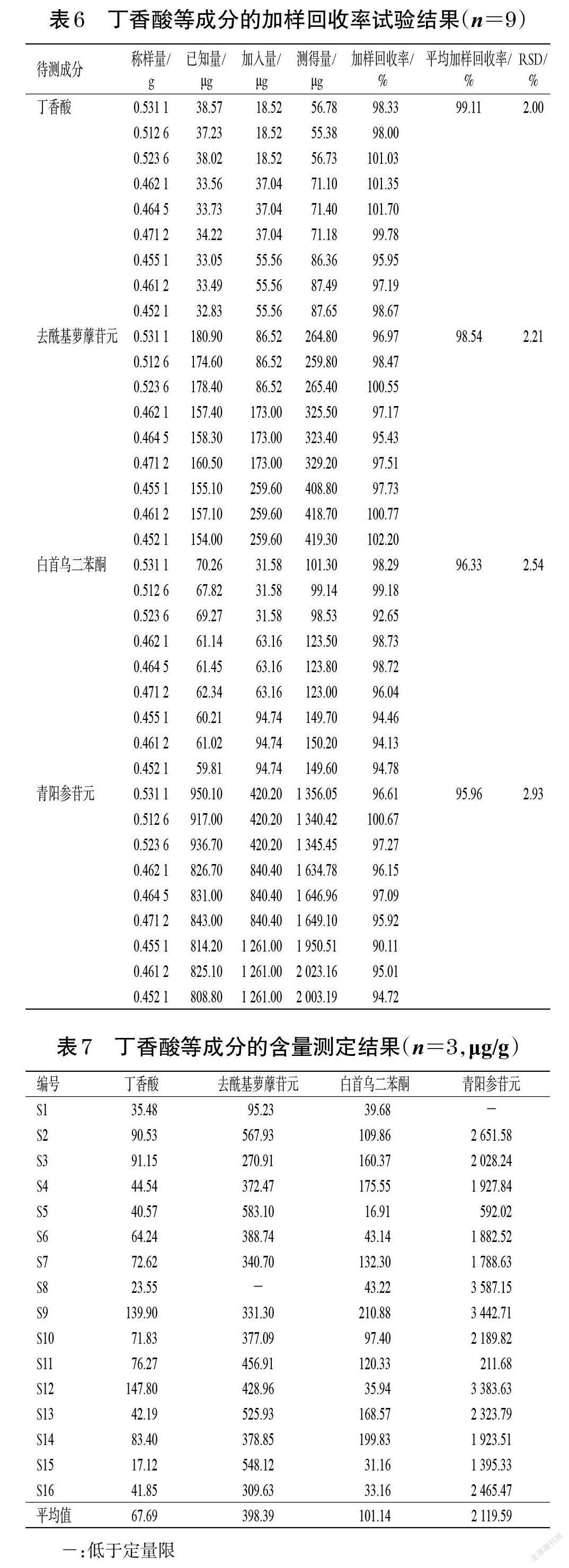
2.5.10 加樣回收率試驗(yàn) 稱(chēng)取已知含量的隔山消藥材(編號(hào)S7)粉末,每份約0.5 g,共9份,按已知量的0.5、1、1.5倍精密加入與待測(cè)成分含量相當(dāng)?shù)幕旌蠈?duì)照品溶液(按“2.5.2”項(xiàng)下方法配制),按“2.5.3”項(xiàng)下方法制備供試品溶液,再按“2.5.1”項(xiàng)下色譜條件進(jìn)樣測(cè)定,記錄峰面積并計(jì)算加樣回收率,結(jié)果見(jiàn)表6。
2.5.11 樣品含量測(cè)定 取16批隔山消藥材粉末約1.0 g,分別按“2.5.3”項(xiàng)下方法制備供試品溶液,再按“2.5.1”項(xiàng)下色譜條件進(jìn)樣測(cè)定,記錄峰面積并按外標(biāo)法計(jì)算樣品含量。每樣品平行測(cè)定3次,取平均值,結(jié)果見(jiàn)表7。
3 討論
3.1 提取溶劑及提取方法的選擇
在前期預(yù)實(shí)驗(yàn)中,本課題組分別考察了不同提取溶劑(水、30%乙醇、50%乙醇、70%乙醇、乙醇、30%甲醇、50%甲醇、70%甲醇、甲醇)對(duì)隔山消藥材指紋圖譜的影響。結(jié)果發(fā)現(xiàn),當(dāng)以30%乙醇為提取溶劑時(shí),各成分色譜峰的響應(yīng)和峰形均較好,基線較平穩(wěn),故選擇30%乙醇為提取溶劑。崔樂(lè)偉[19]采用HPLC法測(cè)定了不同批次斷節(jié)參中青陽(yáng)參苷元含量,當(dāng)回流提取溶劑中加入鹽酸時(shí),青陽(yáng)參苷元等活性成分的色譜響應(yīng)更強(qiáng)。基于此,本研究參考上述文獻(xiàn)的方法,考察了提取溶劑中加鹽酸與不加鹽酸對(duì)各成分色譜響應(yīng)的影響。結(jié)果顯示,與不加鹽酸相比,在提取溶劑中加入0.1 mol/L鹽酸后,各成分的色譜響應(yīng)更強(qiáng)且峰形較好,故最終選擇提取溶劑為0.1 mol/L鹽酸和30%乙醇。本課題組又對(duì)不同提取時(shí)間(0.5、1.0、2.0、3.0、4.0、5.0 h)、提取方法(加熱回流法、超聲提取法)進(jìn)行了考察,結(jié)果顯示,加熱回流4 h時(shí)的提取效果較好且操作簡(jiǎn)單。
3.2 色譜條件的考察
本課題組前期預(yù)實(shí)驗(yàn)考察了不同色譜系統(tǒng)(甲醇-水、甲醇-0.05%磷酸溶液、甲醇-0.1%甲酸溶液、乙腈-水、乙腈-0.05%磷酸溶液、乙腈-0.1%磷酸溶液)、不同洗脫程序、不同柱溫(30、40、45 ℃)、不同流速(0.8、1.0、1.2 mL/min)對(duì)指紋圖譜的影響。結(jié)果顯示,采用本研究所用色譜條件時(shí),所得色譜圖中的色譜峰數(shù)量較多、分離度較好且基線平穩(wěn)。通過(guò)在210~280 nm波長(zhǎng)下對(duì)隔山消藥材進(jìn)行掃描后發(fā)現(xiàn),在210 nm波長(zhǎng)處所得色譜圖基線較平穩(wěn)、出峰信息量大、峰形較好,故選擇210 nm為檢測(cè)波長(zhǎng)。
3.3 指紋圖譜及化學(xué)模式識(shí)別分析
16批隔山消藥材共有29個(gè)共有峰。相似度評(píng)價(jià)結(jié)果顯示,僅S1樣品的相似度為0.723,其余15批樣品的相似度均大于0.950,表明除S1外,其余15批隔山消藥材的質(zhì)量較穩(wěn)定、化學(xué)成分種類(lèi)的差異較小。聚類(lèi)分析結(jié)果顯示,16批隔山消藥材可聚為3類(lèi),其中S1聚為一類(lèi),S3聚為一類(lèi),S2、S4~S16聚為一類(lèi);該結(jié)果與主成分分析結(jié)果一致,表明S1、S3樣品與其余14批樣品存在差異,可能與這兩批藥材的采收時(shí)間、地點(diǎn)、加工方式等不同有關(guān)。正交偏最小二乘-判別分析結(jié)果顯示,20號(hào)峰、10號(hào)峰、25號(hào)峰、12號(hào)峰、15號(hào)峰(青陽(yáng)參苷元)、21號(hào)峰、14號(hào)峰、16號(hào)峰、26號(hào)峰、22號(hào)峰、17號(hào)峰對(duì)應(yīng)的成分是影響隔山消藥材質(zhì)量的差異性成分。
3.4 含量測(cè)定結(jié)果分析
雖然丁香酸、白首烏二苯酮、去酰基蘿藦苷元不是影響隔山消藥材質(zhì)量的差異性成分,但根據(jù)本課題組前期研究結(jié)果,上述化合物為隔山消藥材水溶性部位的主要成分,具有促進(jìn)胃腸道胃動(dòng)素和胃泌素釋放、抑制血管活性腸肽和腫瘤壞死因子α分泌、增加腸道蠕動(dòng)、興奮胃腸平滑肌、促進(jìn)胃排空和腸推進(jìn)、改善功能性消化不良的作用[9]。因此筆者認(rèn)為,這3種成分也可作為隔山消藥材質(zhì)量控制的主要指標(biāo)成分。含量測(cè)定結(jié)果顯示,丁香酸、去酰基蘿藦苷元、白首烏二苯酮、青陽(yáng)參苷元的含量分別為17.12~147.80、95.23~583.10(S8低于定量限)、16.91~210.88、211.68~3 587.15(S1低于定量限)μg/g,各成分含量存在差異,分析其原因可能與不同產(chǎn)地、不同來(lái)源的隔山消藥材的栽培環(huán)境、采收期、種植模式、加工儲(chǔ)存條件等不同有關(guān)。
綜上所述,所建HPLC指紋圖譜和含量測(cè)定方法穩(wěn)定、可靠、準(zhǔn)確度高且重復(fù)性好,結(jié)合化學(xué)模式識(shí)別分析可用于隔山消藥材的質(zhì)量控制。
參考文獻(xiàn)
[ 1 ] 陳艷,徐必學(xué),曹佩雪,等.隔山消化學(xué)成分的研究[J].天然產(chǎn)物研究與開(kāi)發(fā),2013,25(11):1522-1524.
[ 2 ] 王新婕,李振麟,錢(qián)士輝,等.耳葉牛皮消中C21甾體類(lèi)成分研究進(jìn)展[J].中國(guó)野生植物資源,2018,37(3):51-55,63.
[ 3 ] 秦蘭,孫佳,劉春花,等. UPLC-ESI-MS/MS法測(cè)定苗藥隔山消中8個(gè)成分含量[J].貴州醫(yī)科大學(xué)學(xué)報(bào),2021,46(8):886-892.
[ 4 ] 李艷,黎開(kāi)燕.隔山消的藥理作用研究進(jìn)展[J].現(xiàn)代中西醫(yī)結(jié)合雜志,2015,24(2):213-215.
[ 5 ] 吳一振,周明高,周順,等.土家藥隔山消的炮制規(guī)范研究[J].中國(guó)民族醫(yī)藥雜志,2018,24(1):37-39.
[ 6 ] 貴州省藥品監(jiān)督管理.貴州省中藥材、民族藥材質(zhì)量標(biāo)準(zhǔn)[S].貴陽(yáng):貴州科技出版社,2003:384.
[ 7 ] 國(guó)家中醫(yī)藥管理局,《中華本草》編委會(huì).中華本草:苗藥卷[M].貴陽(yáng):貴州科技出版社,2005:283-284.
[ 8 ] 貴陽(yáng)市衛(wèi)生局.貴陽(yáng)民間藥草[M].貴陽(yáng):貴州人民出版社,1959:139.
[ 9 ] 秦蘭,游景瑞,潘潔,等.隔山消提取物對(duì)功能性消化不良模型大鼠腦腸肽及胃腸道功能的作用機(jī)制研究[J].中國(guó)藥業(yè),2021,30(13):23-26.
[10] 謝凱強(qiáng),苑春茂,蹇軍友,等.隔山牛皮消、耳葉牛皮消和戟葉牛皮消中四個(gè)C21-甾體苷元含量比較研究[J].天然產(chǎn)物研究與開(kāi)發(fā),2018,30(2):261-267.
[11] 曹雨,李金花,劉海波,等.隔山消抗神經(jīng)氨酸酶活性及化學(xué)成分研究[J].中草藥,2017,48(21):4485-4492.
[12] 肖培云,藍(lán)海,李龍星. HPLC法測(cè)定隔山消中沒(méi)食子酸的含量研究[J].大理學(xué)院學(xué)報(bào),2009,8(6):9-11.
[13] 孫佳,趙珊,鄒歡,等. HPLC同時(shí)測(cè)定隔山消中5個(gè)成分的含量[J].藥物分析雜志,2015,35(10):1801-1805.
[14] 韓忠耀,向軍,陳建宇,等.不同產(chǎn)地水冬瓜葉HPLC指紋圖譜建立、化學(xué)模式識(shí)別分析及含量測(cè)定[J].中國(guó)藥房,2021,32(10):1224-1229.
[15] 戚華文,徐鑫,高德嵩,等.吳茱萸HPLC指紋圖譜的建立及基原鑒別研究[J].中草藥,2021,52(14):4341-4347.
[16] 孫立麗,王萌,任曉亮.化學(xué)模式識(shí)別方法在中藥質(zhì)量控制研究中的應(yīng)用進(jìn)展[J].中草藥,2017,48(20):4339- 4345.
[17] 劉敏,曹?chē)?guó)瓊,張仕林,等.杜仲補(bǔ)天素丸的HPLC指紋圖譜建立、化學(xué)模式識(shí)別分析及含量測(cè)定[J].中國(guó)藥房,2021,32(8):961-966.
[18] 梁慧,潘曉君,楊文惠,等.基于UPLC指紋圖譜和一測(cè)多評(píng)法的虎杖藥材質(zhì)量評(píng)價(jià)[J].中國(guó)藥房,2021,32(15):1842-1848.
[19] 崔樂(lè)偉.基于物質(zhì)基礎(chǔ)的斷節(jié)參藥材質(zhì)量標(biāo)準(zhǔn)研究[D].昆明:云南中醫(yī)學(xué)院,2014.
(收稿日期:2021-09-09 修回日期:2021-12-29)
(編輯:陳 宏)
