SnO2質量分數對鋅負極用SnO2/ZnO材料結構及電化學性能的影響
2022-03-29 03:13:54程皓郭偉昌汪滔李錚田忠良
中南大學學報(自然科學版) 2022年2期
關鍵詞:質量
程皓,郭偉昌,汪滔,李錚,田忠良
(中南大學冶金與環境學院,湖南長沙,410083)
以金屬Zn 為負極、堿性溶液為電解液的鋅二次電池具有比能量高、成本低以及安全性好等諸多優點[1-3]。但在反復的充放電過程中鋅負極易產生形變、枝晶生長、鈍化以及自腐蝕等現象,造成鋅二次電池循環壽命較短、穩定性差等問題,從而限制了其實際應用[4-5]。為解決以上問題,研究者們在電極結構設計[6]、活性物質表面改性[7]及電解液添加劑[8]等方面開展了大量研究。
其中,用高析氫過電位的金屬(如Sn,Bi)或其化合物(如SnO2,Bi2O3)對ZnO 進行改性,可有效抑制析氫腐蝕,緩解鋅枝晶生長從而改善電池的電化學性能[9]。此類化合物通常在電池工作過程中優先被還原成金屬,提高電極導電性,改善負極活性物質的沉積過程,抑制鋅負極發生枝晶生長的同時降低其析氫腐蝕速率,提高活性物質利用率[10-11]。ZHANG 等[12]采用水熱法制備出鋅鎳二次電池鋅負極用SnO2/ZnO 復合材料,相較于純ZnO材料,該復合材料顯著提高了電池的循環壽命和循環穩定性,但該復合材料中錫分布不均勻,表面粗糙,形貌不均勻,平均粒徑為80~100 nm,導致其平均放電比容量不高,低于550 mA·h/g。可見,通過SnO2改性ZnO 材料,可在一定程度上提高鋅負極的循環穩定性,提高電池循環壽命,但由于材料形貌及粒徑不均勻,故放電比容量還有待進一步提升。JOHNSON 等[13]通過錫摻雜對ZnO納米顆粒進行改性,發現當錫的質量分數為2%和4%時,材料呈多孔結構,而當錫質量分數從6%增加到10%時,多孔結構消失,平均粒徑變大且出現團聚現象。此外,在鋰離子電池負極用SnO2/ZnO 材料合成過程中,隨著錫質量分數增加,SnO2/ZnO 材料的平均粒徑減小,其電化學性能也得到明顯改善,主要是因為在電池工作中,該材料可保持較好的形貌以維持電池穩定[14-15]。因此,可將納米氧化鋅與無機物表面修飾的方法相結合,從而進一步改善電池的電化學性能。
本文作者利用ZIF-8在一定條件下可得到納米級金屬氧化物的特點,將吸附-還原法與熱處理結合制備ZIF-8 衍生的SnO2/ZnO 材料,對比不同熱處理溫度和時間制備的材料的物相及微觀形貌,以優化熱處理條件,在此基礎上,進一步研究材料中SnO2質量分數對ZIF-8 衍生SnO2/ZnO 材料的物相結構、微觀形貌及其電化學性能的影響,揭示電化學性能與材料理化性質之間的關系。
1 實驗
1.1 Sn@ZIF-8和SnO2/ZnO材料的制備
首先,將一定量(0.02,0.04 和0.06 g)的二水合氯化亞錫(SnCl2·2H2O,國藥集團,分析純)溶于40 mL 甲醇(CH3OH,國藥集團,分析純)中,加入0.5 g ZIF-8,超聲處理30 min 后轉移至水浴鍋中,室溫下攪拌12 h得到懸濁液。
然后,將1.32 mmol的硼氫化鈉(NaBH4,國藥集團,分析純)溶于5 mL去離子水后緩慢加入上述懸濁液中,攪拌30 min 后真空抽濾,濾餅用CH3OH 和去離子水洗滌3 次后,于65 ℃的鼓風干燥箱中干燥12 h,得到Sn@ZIF-8前驅體。
最后,將Sn@ZIF-8 研磨成粉末后在空氣中以4 ℃/min 升至目標溫度,并保溫一定時間,得到SnO2/ZnO材料。作為比較,將ZIF-8在相同條件下熱處理得到純氧化鋅材料(純ZnO)。
1.2 材料表征
所制備的材料采用熱重分析儀(TGA,SDTQ600)分析前驅體的熱性能;采用X 射線衍射儀(XRD,Rigaku3014)和電感耦合等離子體原子發射光譜法(ICP-OES,iCAP7200 Radial)分析材料的物相組成和元素質量分數;采用掃描電子顯微鏡(SEM,JSM-6360LV)表征材料的微觀結構。
1.3 電化學性能測試
按質量比為8∶1∶1將SnO2/ZnO、導電碳和聚四氟乙烯乳液(PTFE,阿拉丁,60%)混合并涂敷于銅網上(正方形網格邊長×邊長為1 cm×1 cm),用雙軸輥壓機壓至0.3 mm左右,于60 ℃下干燥12 h得到鋅負極。以燒結鎳為正極,溶有0.5 mol/L 氧化鋅(ZnO,西隴科學,分析純)的6.0 mol/L 氫氧化鉀(KOH,國藥集團,分析純)溶液為電解液(溶劑為去離子水),并分別在正負極包裹一層聚丙烯微孔隔膜組裝成鋅鎳電池。采用LANDE電池檢測系統(CT2001A)對電池進行恒電流充放電測試,在659 mA/g 電流密度下恒電流充電1 h,并在相同電流密度下放電至1.2 V終止(標稱比容量為659 mA·h/g)。采用三電極體系(以制備的SnO2/ZnO或純ZnO為工作電極,燒結鎳電極為對電極,Hg/HgO電極為參比電極)通過電化學工作站(CHI660E,A17177)進行循環伏安性能測試。
2 熱處理工藝優化
對比熱處理溫度和熱處理時間對材料微觀結構的影響,以優化熱處理工藝。圖1所示為熱處理溫度對材料物相的影響。由圖1(a)可見:Sn@ZIF-8前驅體在400 ℃質量率損失約為8.8%,主要為材料中結晶水的損失,說明前驅體在400 ℃之前保持穩定;當溫度高于400 ℃,質量損失急劇增加,表明材料開始劇烈分解;超過600 ℃后,質量損失率為61.78%,材料質量逐漸穩定,說明材料中的有機物已揮發,故要得到最終產物,熱處理溫度應控制在600 ℃以上。從圖1(b)可見:在不同溫度下得到的材料均出現ZnO 和SnO2的特征峰,說明均成功制備出SnO2/ZnO 材料,但對比發現900 ℃熱處理后的材料的XRD 圖中還出現了Zn2SnO4的特征峰,是因為ZnO 和SnO2在900 ℃下會發生反應(1),導致新物相生成,該雜質相降低了目標產物中錫的質量分數,因此,在900 ℃不利于SnO2/ZnO材料的制備。

圖1 熱處理溫度對材料物相的影響Fig.1 Influence of heat treatment temperature on material phase

采用Scherrer方程[16-17]估算樣品的平均粒徑:

式中:D為晶粒垂直于晶面方向的平均厚度;k為Scherrer 常 數(k=1.075);λ為X射線波長(λ=0.154 056 nm);β為半峰寬度;θ為衍射角。計算得到700 ℃和800 ℃熱處理后SnO2/ZnO 材料的平均粒徑分別為29.5 nm和22.4 nm。
圖2 所示為不同熱處理溫度得到的SnO2/ZnO材料的SEM 圖,由圖2 可見:由于ZnO 在高溫下會向致密化發展,且隨溫度升高,顆粒粒徑變小[18-19],故觀察到800 ℃處理后的材料顆粒粒徑比在700 ℃下處理的材料的顆粒粒徑小(圖2),這與計算結果一致。而SnO2/ZnO材料粒徑大導致SnO2對ZnO 的修飾不充分,ZnO 在電解液中溶解快,活性物質的利用率降低[11],所以,選擇800 ℃為最佳熱處理溫度。

圖2 不同熱處理溫度得到的SnO2/ZnO材料的SEM圖Fig.2 SEM images of SnO2/ZnO materials heat-treated at different temperatures
圖3 所示為不同熱處理時間得到的SnO2/ZnO材料的XRD 圖。由圖3 可知:在不同熱處理時間下得到的材料均出現了ZnO 和SnO2的特征峰,說明均成功制備出SnO2/ZnO材料,但在熱處理5 h的XRD圖中還存在Zn2SnO4的特征峰,說明熱處理時間較短時,材料中存在雜相,不利于SnO2/ZnO 材料的制備。

圖3 不同熱處理時間得到的SnO2/ZnO材料的XRD圖Fig.3 XRD patterns of SnO2/ZnO materials with different heat treatment time
根據式(2)計算得熱處理7 h 和9 h 的SnO2/ZnO材料的平均粒徑分別為23.7 nm 和22.4 nm。分析微觀形貌,對比800 ℃處理7 h和9 h得到的SnO2/ZnO材料的SEM 圖(圖4),發現熱處理7 h 的SnO2/ZnO材料存在粒徑較大且形狀不規則的顆粒,隨著熱處理時間延長,熱處理9 h 得到的材料顆粒形貌、分布均勻,因此,最優的熱處理時間為9 h。

圖4 不同熱處理時間得到的SnO2/ZnO材料的SEM圖Fig.4 SEM images of SnO2/ZnO materials with different heat treatment time
3 SnO2質量分數對SnO2/ZnO 材料結構及電化學性能的影響
3.1 SnO2質量分數對材料結構的影響
由ICP-OES測試結果計算可得,在SnCl2·2H2O添加量分別為0.02,0.04 和0.06 g 時制備的SnO2/ZnO 材料中,SnO2質量分數分別為7.7%,15.1%和23.0%,將其分別記作Sn-1,Sn-2 和Sn-3。圖5(a)所示為不同SnO2質量分數的SnO2/ZnO 材料的XRD圖,圖5(a)中均出現了ZnO和SnO2的特征峰,且隨著SnO2質量分數增加,SnO2特征峰的強度增強。通過式(2)計算可得SnO2質量分數為7.7%,15.1%和23.0%的SnO2/ZnO材料的平均粒徑分別為24.8,22.4和23.0 nm。圖5(b),(c)和(d)所示分別為Sn-1,Sn-2和Sn-3的SEM圖,由于Sn-1中SnO2質量分數較小,部分顆粒相互接觸,空間位阻作用弱,氧化鋅顆粒發生團聚和生長,致使部分顆粒粒徑較大,所以,圖5(b)中存在粒徑較大的顆粒;材料Sn-2的粒徑均勻;而錫質量分數較高時也會阻礙顆粒向致密化發展,發生團聚,所以,Sn-3中還存在少量大顆粒物質。可見,SnO2質量分數為15.1%的SnO2/ZnO材料具有更均勻的粒徑和形貌。

圖5 不同SnO2質量分數的SnO2/ZnO材料的XRD圖及SEM圖Fig.5 XRD patterns and SEM images of SnO2/ZnO materials with different mass fractions of SnO2
3.2 SnO2質量分數對材料電化學性能的影響
對不同SnO2質量分數的SnO2/ZnO材料進行循環伏安測試,結果如圖6和表1所示。由圖6可見:隨著SnO2質量分數增加,陰極峰和陽極峰的位置沒有發生明顯偏移,說明SnO2的存在沒有改變鋅負極的反應機理。循環伏安曲線中-1.24 V 左右出現陽極峰,在該電位下生成鋅酸根離子,如式(3)所示:

SnO2的存在限制了活性物質與電解液的接觸,同時,電解液中OH-擴散產生了圖6 中-1.13 V 左右的陽極峰。

圖6 純ZnO和不同SnO2質量分數的SnO2/ZnO材料的循環伏安曲線Fig.6 Cyclic voltammetry profiles of pure ZnO and SnO2/ZnO nanocomposites with different mass fractions of SnO2

而-1.53 V 左右的陰極峰則為充電過程還原反應:
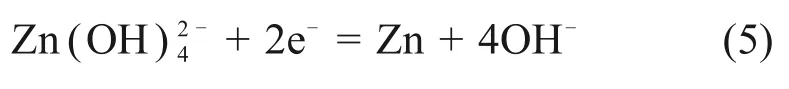
由于SnO2的還原電位比ZnO 的還原電位高,所以,在活化階段會被優先還原為金屬錫,而錫具有較高析氫過電位,會進一步抑制負極的析氫腐蝕,有利于增強ZnO在堿性電解液中的穩定性[12]。
循環伏安曲線中陰極峰和陽極峰之間的電壓差反映了電極反應的可逆程度,電壓差小表示電極反應可逆程度高,極化小[20]。據表1 可知:Sn-2材料具有更小電壓差(0.281 V),即可逆程度高。雖然錫會改善材料的導電性,但過量錫會阻礙OH-與活性物質接觸,增大極化,因此,Sn-3 材料陰極峰與陽極峰之間的電壓差(0.289 V)較Sn-2 的電壓差(0.287 V)大。

表1 純ZnO和不同SnO2質量分數的SnO2/ZnO材料的循環伏安曲線數據Table 1 Data for cyclic voltammetry profiles of pure ZnO and SnO2/ZnO nanocomposites with different mass fraction of SnO2
圖7(a)為純ZnO 和不同SnO2質量分數的SnO2/ZnO 材料的循環性能和充放電曲線,表2 所示為不同材料在第50,100,129 和200 圈的放電比容量。由于鋅負極存在枝晶、形變以及腐蝕等問題,故純ZnO 的放電比容量低且波動大。而Sn-1材料在前50 圈較穩定,后續衰減較快,平均放電比容量為519 mA·h/g。Sn-2 和Sn-3 材料的循環穩定性強,但Sn-3 材料中錫質量分數較Sn-2 錫質量分數高,限制了活性物質與電解質的有效接觸,故Sn-2材料的容量及容量保持率更高,平均放電比容量為600 mA·h/g,循環200 圈的容量保持率為93.66%。而Sn-3的平均放電比容量為535 mA·h/g,循環200 圈的容量保持率為89.11%。且Sn-2 材料的容量明顯高于已有文獻中材料的容量[12,21-24]。

表2 純ZnO和不同SnO2質量分數的SnO2/ZnO材料在不同放電圈數的放電比容量Table 2 Specific discharge capacity of pure ZnO and SnO2/ZnO nanocomposites with different mass fractions of SnO2 in different discharge cycles

圖7 純ZnO和不同SnO2質量分數的SnO2/ZnO材料的循環性能和充放電曲線Fig.7 Cycle performances and galvanostatic charge-discharge profiles of pure ZnO and SnO2/ZnO nanocomposites with different contents of SnO2
為進一步研究不同SnO2質量分數的SnO2/ZnO充放電性能,對其在第50 圈時的充放電曲線進行分析,如圖7(b)所示。由圖7(b)可知:純氧化鋅電極的中值電壓差最大,即極化最大,而由于適量錫可以有效抑制負極的析氫腐蝕并改善材料導電性,所以,Sn-2 電極的充放電曲線的中值電壓差最小(0.134 5 V),即電極極化小,這與循環伏安曲線所反映的結論一致。
4 結論
1)以ZIF-8 為載體,將吸附-還原法與熱處理相結合,制備了SnO2/ZnO 材料并將其用作鋅鎳電池負極。在熱處理溫度為800 ℃,熱處理時間為9 h時,所制備的材料物相單一,形貌均勻。
2) SnO2質量分數對SnO2/ZnO 材料顆粒分布、粒度及電化學性能有一定的影響,隨著材料中SnO2質量分數從7.7%增加至15.1%,顆粒粒徑減小且分布均勻,第50 圈充放電曲線的中值電壓差從0.157 8 V 減小至0.134 5 V,平均放電比容量從519 mA·h/g 增加至600 mA·h/g,極化變小,循環穩定性及放電比容量提升;當SnO2質量分數從15.1%增加至23.0%時,顆粒粒徑變大,且分布不均,第50 圈充放電曲線的中值電壓差從0.134 5 V減小至0.138 3 V,平均放電比容量從600 mA·h/g減小至535 mA·h/g,極化增大,循環穩定性變差,放電比容量降低。
3)SnO2質量分數為15.1%的SnO2/ZnO 顆粒粒徑最小,分布均勻,鋅鎳電池負極在659 mA/g的電流密度下循環200圈的放電比容量為562.0 mA·h/g,容量保持率為93.66%,表現出最小極化、最高放電容量及最穩定循環性能。
猜你喜歡
中學生數理化·中考版(2022年10期)2022-11-10 09:37:42
中學生數理化·八年級物理人教版(2022年12期)2022-02-14 07:08:42
中學生數理化·八年級物理人教版(2021年12期)2021-12-31 03:23:08
中學生數理化·中考版(2020年10期)2020-11-27 01:59:48
中國生殖健康(2019年2期)2019-08-23 08:12:08
石油化工建設(2018年6期)2018-04-22 03:16:54
產品可靠性報告(2017年7期)2017-09-05 09:49:12
中學生數理化·八年級物理人教版(2017年12期)2017-04-18 12:59:38
汽車觀察(2016年3期)2016-02-28 13:16:26
民生周刊(2014年7期)2014-03-28 01:30:54
