應激顆粒在細胞衰老中的作用
2022-04-13 05:11:00金志剛蔣瑩佩張晶晶
浙江師范大學學報(自然科學版) 2022年2期
關鍵詞:信號
金志剛, 蔣瑩佩, 張晶晶
(浙江師范大學 化學與生命科學學院,浙江 金華 321004)
1 細胞衰老及其調控機制
1.1 衰老表型特征
細胞衰老(cellular senescence)是當不斷增殖的細胞受到氧化應激、DNA損傷和癌基因活化等內源或外源壓力時誘發的細胞周期永久停滯的一種狀態[1].對細胞衰老的首次描述可以追溯到20世紀60年代,Hayflick等[2]觀察到在體外培養條件下,人類二倍體成纖維細胞增殖至一定代數后達到極限并停止增殖,這種極限被稱為“Hayflick極限”.細胞衰老會誘發永久的細胞周期停滯,并引發細胞表型變化,如生物活性分泌物的產生,稱為衰老相關的分泌表型(senescence-associated secretory phenotype,SASP)[3].機體在發育過程中通過衰老細胞誘導組織重塑、傷口愈合和腫瘤抑制反應[4-5],這些衰老細胞通過SASP的產生來招募免疫細胞,誘發機體免疫反應,從而清除衰老細胞[6-13].但隨著年齡的增長,機體免疫機能逐漸衰退,此時衰老細胞分泌的SASP則發揮免疫抑制功能[14-15],引發系統性炎癥甚至破壞組織內穩態[16-19],成為許多衰老相關疾病的誘因.因此,通過選擇性清除衰老細胞來延緩衰老和限制衰老引起功能障礙的方法,即衰老細胞清除療法(senotherapy),目前受到廣泛關注[3,19-20].
具有抑制絲氨酸蛋白酶活性的纖溶酶原激活物抑制子-1(plasminogen activator inhibitor-1,PAI-1)是參與SASP的一種重要分泌蛋白,最早在1991年發現,過早衰老的細胞中PAI-1表達升高[21],此后陸續在其他組織發現其與衰老細胞緊密聯系,被認為是衰老標志物之一[3,22-23].靶向PAI-1的小分子抑制劑TM5441可以有效防止阿霉素誘導的心肌細胞、成纖維細胞和內皮細胞的衰老[24-26],從而改善小鼠的血管衰老及細胞衰老引起的損傷[27-28].除此之外,衰老表型特征還包括但不限于衰老相關的β-半乳糖苷酶活性(SA-β-gal)增加,p16INK4A、p53和p21等細胞周期抑制因子表達水平增高,γ-H2AX等的DNA損傷,衰老相關異染色質灶(senescence-associated heterochromatin foci,SAHF)等,這些衰老表型特征不僅可作為評估各種衰老模型的衰老進程標志物[29],還可作為衰老細胞清除療法的藥物靶點[30-33].
1.2 調控細胞衰老的信號通路
過去30年對細胞衰老的研究已經從衰老表型轉向這些表型背后的分子機制.目前通過遺傳分析發現,調控細胞衰老的信號通路主要涉及營養攝取和蛋白質穩態等方面(見圖1).
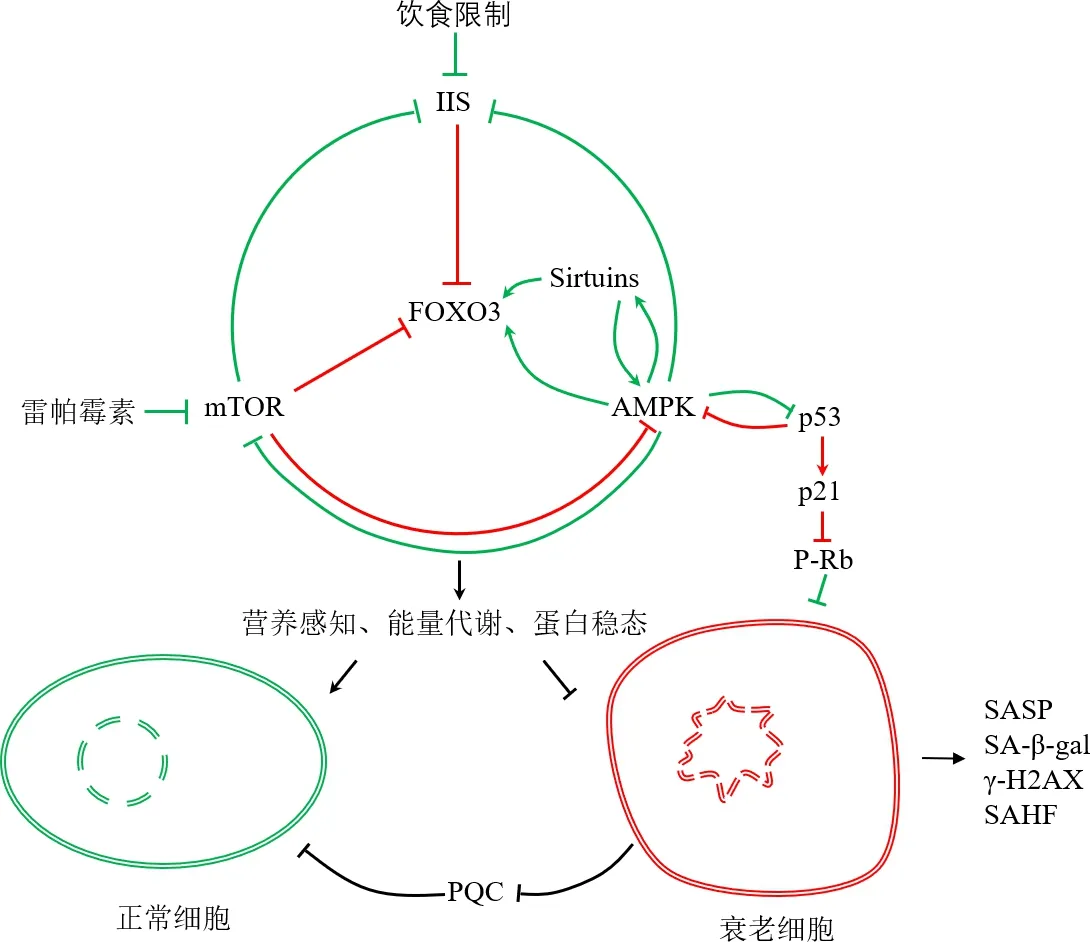
紅色表示促進細胞衰老;綠色表示維持正常細胞,抑制細胞衰老
IIS通路、mTOR通路、AMPK、FOXO和sirtuins是調控細胞衰老的主要途徑,通過調節細胞營養感知、能量代謝和蛋白穩態來拮抗調控細胞衰老,且在酵母、線蟲、蒼蠅和哺乳動物中是相對保守的[3,34-35],以下作簡要概述:1)從營養感知來說,機體衰老與胰島素/IGF-1信號通路(insulin/IGF-1signaling,IIS)有關.營養匱乏時,IIS通路被阻斷,導致叉頭盒蛋白O(the fork head box protein O,FOXO)線蟲同源蛋白DAF-16活化,進而激活了下游抗衰老靶基因的轉錄[35-38],表明降低IIS通路活性可以延長線蟲壽命.除此之外,IIS通路還會引起葡萄糖運輸和脂肪代謝受損,而抗衰老因子則通過降低細胞對IGF-1的利用、激活腺苷單磷酸激活激酶(AMP-activated kinase,AMPK)、阻斷哺乳動物雷帕霉素靶蛋白(mammalian target of rapamycin,mTOR)通路、增加自噬活性和維持線粒體完整性來促進機體長壽[35].2)mTOR通路,感知營養的另一個重要途徑,可以調節細胞生長,整合胰島素信號和細胞應激信號,從而調節mRNA翻譯、自噬、轉錄和線粒體功能等過程[35,38].激活mTOR復合物1(target of rapamycin complex1,TORC1)會加速細胞和組織衰老的過程,而通過雷帕霉素、TORC1基因失活或飲食限制(dietary restriction,DR)來抑制mTOR通路,可以延長模式生物壽命并改善一系列生理表現[39].3)AMPK是細胞內重要的能量感受器和調節器,可以感知細胞壓力、運動、多種激素及影響細胞代謝的物質等各種刺激.AMPK在細胞能量水平低時被激活,并通過抑制TORC1阻斷蛋白質合成,通過促進線粒體呼吸維持細胞能量[35,40],從而延緩細胞衰老.4)NAD+是三羧酸循環和電子呼吸鏈上重要的輔酶,NAD+依賴性蛋白去乙酰化酶sirtuins家族蛋白是重要的衰老調節因子家族.早在對酵母菌、果蠅和線蟲的研究中發現,sirtuins家族顯著延長了這些模式生物的壽命[41-42].sirtuins家族的7個成員(SIRT1-7),通過調控p53,AMPK等信號通路[43-44],來調節線粒體穩態、基因組穩定性、NF-κB信號傳導和葡萄糖代謝,參與DR介導延長細胞壽命[41,45-46].綜上,IIS通路、mTOR通路、AMPK和sirtuins通過對營養或能量的感知和調節,在細胞衰老過程中發揮了重要作用.
1.3 衰老細胞的蛋白質穩態失衡
蛋白質穩態失衡既是細胞衰老的重要特征,也是誘發機體衰老的主要原因[47-48].細胞蛋白質穩態的維持需要精確控制蛋白質合成、折疊、構象維持和降解等過程,這些過程需要不同類別的分子伴侶及其相關調節因子,以及蛋白酶體或溶酶體共同協調,同時還需要ATP供能,來減少蛋白錯誤折疊或聚集[49-51].隨著年齡的增長,熱休克蛋白HSP70(heat-shock protein 70)[52-53]和熱休克轉錄因子HSF-1(heat shock transcription factor-1)[54]等分子伴侶活性下降,蛋白酶體功能減弱,導致包括內質網的未折疊蛋白反應(unfolded protein response,UPR)在內的蛋白質量控制系統(protein quality-control system,PQC)嚴重受損,PQC受損導致的蛋白質聚集是衰老細胞及衰老相關疾病的重要標志[48-49,51,55-57].在許多模型中,通過外源性表達單個分子伴侶或通過藥理學誘導應激反應上調多個分子伴侶,已被證明可以防止毒性聚集體的形成或誘導形成毒性較小的聚集體[49,58-59].這種PQC成分的上調允許細胞清除急性壓力誘導的錯誤折疊蛋白質及其聚集體,但無法清除與神經退行性疾病相關的淀粉樣蛋白質聚集體,如β-淀粉樣蛋白、Tau、α-突觸核蛋白、超氧化物歧化酶1(superoxide dismutase 1,SOD1)和TDP-43等神經毒性蛋白質聚集體[49,60].衰老神經元內蛋白酶體和HSP70活性下降,會促進TDP-43液滴形成,當ATP水平降低時,TDP-43液滴進而向固態聚集體轉變[61].這些蛋白質聚集體可以通過衰老細胞清除療法來清除,從而延緩衰老相關疾病的表型[62-63].這些證據都表明,蛋白質穩態失衡對細胞衰老影響甚大,因此,闡明細胞衰老過程中的蛋白質穩態失衡可為抵抗衰老及治療衰老相關疾病提供重要依據.
2 應激顆粒簡介
如前所述,IIS通路、mTOR通路、AMPK和sirtuins作為營養或能量的感知器和調節器,在細胞衰老過程中發揮了重要作用.近年研究發現,應激顆粒(stress granules,SGs)作為壓力的感受器和調節器,也參與調控了細胞衰老.真核細胞在面對熱刺激、病毒感染、高滲透壓、氧化應激及紫外輻射等不利環境條件時有一種典型的應激反應,包括抑制整體翻譯及選擇性上調分子伴侶[64-65].處于翻譯暫停狀態的多聚體核糖體解離,導致細胞質中無核糖體結合的游離mRNA濃度升高,并與一些RNA結合蛋白(RNA-binding proteins,RBPs)結合,最終形成了細胞質凝聚體,即SGs.SGs主要由mRNA-蛋白復合物組成,其蛋白組分包括多種翻譯起始因子、RBPs和非RBPs[65-66].
SGs是動態復雜可變的生物分子凝聚體,其組成和結構在不同類型的壓力下會發生巨大變化,這種多樣性被認為是依賴于SGs內部蛋白質和RNA之間的不同相互作用,并反映了細胞對各種環境壓力快速響應的能力[65,67].因此,SGs的動態過程及其調控,對于細胞應對應激壓力就顯得至關重要.
SGs是無膜細胞器,具有成分、功能和動力學各不相同的2層結構,即由松散相互作用的分子形成的外殼及SGs核心與mRNA緊密結合構成的內核[64-65,68].由多價弱大分子相互作用(蛋白質-蛋白質、蛋白質-RNA和RNA-RNA相互作用)驅動的液-液相分離(liquid-liquid phase separation,LLPS)是無膜細胞器自組裝的一個重要條件[65,69].LLPS是將混合溶液中的成分分離成2種不同相的過程,某些成分在一個相中富集,在另一個相中減少[70],使得這2個相中的成分可以很容易地重新排列,并可以快速地相互交換物質.這也是SGs這類無膜細胞器既能夠與胞漿保持分離,又能對環境壓力作出快速反應的原因之一[69].高濃度的大分子必須達到臨界閾值才能啟動LLPS,但是超過一定的濃度,SGs內部的大分子如蛋白質,更傾向于形成聚集體.這可能導致LLPS產生的液體組分具有凝膠狀或固體狀的特性,從而使得液體組分無法正常工作,如病理性的胞質或核蛋白質聚集體[66,69,71].
SGs的一個重要功能是使細胞能夠適應不同的環境變化.在壓力條件下,SGs為細胞的關鍵組分提供了暫存場所,如暫停翻譯的mRNA和暫停信號傳遞的信號轉導分子,從而為適應或消除壓力后mRNA返回多聚核糖體重啟翻譯、信號轉導分子返回胞漿重啟信號轉導等正常生理活動的恢復奠定了基礎.通過壓力誘導的SGs組裝來招募信號分子,通過壓力消除后的SGs解聚來釋放信號分子,SGs因而也成了細胞應激反應的信號轉導中心.比如SGs與mTOR信號通路存在互相調控.壓力刺激后,TORC1定位至SGs被隔離,信號終止;而在壓力消除后,TORC1隨著SGs解聚被釋放并重新激活信號[72];TORC1則通過磷酸化4E-BP1(4E-binding protein 1)驅動eIF4E介導的SGs形成[73].此外在亞砷酸鹽(arsenite,AS)誘導下,TORC1組分定位在SGs中從而抑制氧化應激誘導的細胞凋亡[74].研究表明,SGs通過招募促凋亡信號分子RACK1(the receptor for activated C kinase)抑制細胞凋亡[75-76].
綜上,在整個應激過程中,SGs通過在壓力條件下臨時儲存mRNA和信號分子,暫停了mRNA翻譯和信號轉導,并允許壓力消除后隨著SGs解聚快速重啟翻譯和信號轉導.因此,SGs在應激反應中發揮了重要功能,而SGs的異常也導致了mRNA和蛋白質等在內的生物大分子的功能紊亂,并與多種衰老人群易感疾病相關,包括神經退行性疾病和腫瘤等.目前越來越多的研究表明,SGs與細胞衰老存在著密切關聯(見圖2).
3 應激顆粒對細胞衰老的調控
3.1 應激顆粒調控衰老相關信號通路
當細胞受到外界刺激時,會快速響應應激反應,而SGs作為信號轉導中心,通過招募支架蛋白、轉錄翻譯相關蛋白質、信號通路中各種相關激酶及重要成員,從而調控細胞適應環境變化.當細胞無法承受外界刺激時,會啟動細胞衰老、退出細胞周期,限制舊細胞或受損細胞的復制,從而保證周圍正常細胞的存活.同樣,細胞衰老也是對細胞應激的一種響應,目前已有許多證據表明,SGs與細胞衰老密切相關,二者相互影響和調控(見圖2).

圖2 SGs與細胞衰老相互調控
SGs通過招募衰老相關因子調控細胞衰老.1)PAI-1是一種衰老激活因子,正常條件下以分泌蛋白的形式分布于細胞外基質.當細胞受到AS刺激后,PAI-1被招募至SGs,SGs抑制了PAI-1的促衰老功能,從而促進了細胞周期蛋白D1(cyclin D1)入核及Rb磷酸化,使得細胞維持增殖、非衰老狀態[77].另外,CO誘導的SGs可以通過隔離PAI-1抑制博來霉素誘導的細胞衰老,而同時加入SGs抑制劑ISRIB處理后,SGs組裝被抑制,使得博來霉素誘導的細胞衰老被重啟,這提示SGs可以通過招募衰老關鍵因子致其功能失活,從而對抗細胞衰老[78].2)TORC1被過度激活能促進細胞衰老,而在應激條件下,TORC1被隔離在SGs,使得mTOR信號終止,延緩細胞衰老.當壓力消退后隨著SGs解聚,TOPC1被釋放,才重新激活mTOR信號[72].除此之外,線粒體功能及生物發生、基因組穩態[35,79-81]等方面的證據提示,SGs在細胞響應外界刺激時形成的RNA和蛋白網絡,同樣也可能是細胞無法響應外界刺激時進入衰老過程的信號轉導中心.
綜上,SGs作為細胞應激反應的臨時信號轉導中心,賦予細胞應激反應能力,并通過招募衰老相關調控因子,發揮延緩細胞衰老的作用.
3.2 異常應激顆粒對衰老相關疾病的影響
正常情況下,SGs的產生是為了保護細胞不受急性應激的損害,即使有異常的SGs產生也會在PQC的調控下被靶向蛋白酶體或溶酶體清除,但在病理條件下,異常SGs會是某些疾病的病灶.SGs的形成和細胞衰老這兩個過程有許多相似的特點,尤其是蛋白質穩態變化,因此,病理條件下SGs也可能是衰老相關疾病的病灶.目前發現,細胞年齡相關性蛋白在各種組織中出現多種生理和病理蛋白質聚集體,尤其在神經退行性疾病中發現多種蛋白質在衰老過程中極易形成聚集體[79].
已經有證據顯示,SG蛋白PAB-1和TIAR-2均能在衰老的線蟲中形成聚集體,這表明SGs蛋白質聚集體可能加速衰老,減少壽命[82-83].另外,SGs所包含的RBPs大多含有內在無序區,使得這些蛋白構象靈活、易于折疊且傾向于自發轉變為不溶性的聚集體,這種特性使得與之互作的其他蛋白和RNA也傾向于聚集,從而形成不可逆的聚集體,尤其當這些RBPs在特定區域發生突變時,更容易形成聚集體.這些異常的、不可逆的聚集體在去除應激后會導致SGs持續性存在,進而引起細胞內的其他病理變化,這其中就可能涉及某些衰老調控因子[65-66,69-71].
此外,一些包括TDP-43,FUS和Ataxin-2在內的胞質淀粉樣蛋白質聚集體能通過隔離核質轉運體來干擾核完整性和核質運輸,這些影響可能加速衰老和神經退行性疾病的發病[84-88].而這些胞質淀粉樣蛋白質聚集體也被報道與SGs有密切聯系[66,85],因此,闡明SGs的動態過程及異常發生,對衰老相關疾病的研究及治療有重要意義.
4 細胞衰老對應激顆粒的動態調控
4.1 細胞衰老參與應激顆粒動態過程調控
細胞衰老本意是保護細胞以免獲得不必要的損害,但隨著衰老進程的繼續,基因組穩態失衡、端粒損耗、蛋白質穩態喪失、營養感知失調、線粒體功能障礙等影響細胞的生存,從而導致衰老成為誘發許多疾病的主要危險因素,包括癌癥、神經退行性疾病和心血管疾病[47,64].目前已有證據表明,伴隨著細胞衰老而來的細胞內環境的變化同樣影響SGs的動態過程(見圖2).
自然狀態下的細胞衰老會隨著細胞增殖逐漸累積不利于細胞生存的環境條件,是一個慢性應激的過程.隨著細胞進入衰老,急性應激條件下仍能形成SGs,但在慢性氧化應激下SGs 的數量會減少甚至無法形成SGs,這是由于在慢性應激下促進SGs解聚的熱休克蛋白HSP70和自噬蛋白ATG5表達上調,導致SGs被大量清除[77,89-90].這些發現表明,衰老細胞能夠根據施加壓力的性質改變SG動態過程,影響SG功能和完整性.
但有趣的是,即使敲低HSP70和ATG5,衰老細胞中慢性應激下SGs數量仍很少,可能還有其他通路影響SGs的形成或清除.Moujaber等[91]的工作則證明了這一點,他們發現細胞衰老通過下調轉錄因子Sp1和蛋白磷酸酶1的亞基CReP水平,前者調節SGs核心蛋白G3BP1(Ras-GAP SH3-binding protein 1)和TIA-1(T-cell-restricted intracellular antigen-1)豐度,后者抑制eIF2α應激依賴性磷酸化,從而降低G3BP1和TIA-1水平,抑制SGs形成.
盡管衰老細胞對慢性應激和急性應激的響應不同,但它們調節SGs的能力和對應激的適應能力普遍受損.這可以部分解釋為什么衰老細胞從壓力中恢復得比非衰老細胞慢,以及壓力與衰老相關疾病的高度相關性.
4.2 細胞衰老促進應激顆粒組分形成不溶性聚集體
細胞衰老的過程與多種細胞生理活動的失調有關,包括蛋白質穩態失衡、細胞內pH值降低和活性氧增加[92-93].這種細胞內環境的變化在衰老過程中會進一步影響SGs的形成:1)影響蛋白的相分離行為導致改變與其互作蛋白的結合親和性,從而被招募到SGs中,反過來又會影響SGs組成、物理性質和結構組織,包括內在無序蛋白的濃度,使其更傾向于不溶性相轉變,如前述的FUS,TDP-43[69,94].2)包括活性氧在內的氧化應激是公認的SGs主要誘導因素之一,SGs被氧化應激誘導組裝,并在細胞解除或適應應激后解聚,而在持續氧化應激中招募并穩定一些病理性寡聚化的RBPs,促使這些RBPs形成聚集體,如SOD1和Tau[95-96].3)SGs的正常功能發揮需要分子伴侶介導的監測機制,以免SGs持續存在損害細胞正常翻譯.HSPB8-BAG3-HSP70伴侶復合物是SGs組成和動態的關鍵調節劑,然而這種監測機制在細胞衰老過程中,由于HSP70活性下降被破壞,使得錯誤折疊的蛋白質和有缺陷的核糖體產物就會在SGs中積累,并隨著有缺陷的SGs解聚引發異常的相分離[97-98].4)SGs的組裝、解聚和清除都需要消耗能量,因此,伴隨細胞衰老的線粒體功能障礙及營養感知失調很有可能損害SGs的動態過程,甚至可能引起SGs或其中某些與疾病相關蛋白的異常相分離[99].
有證據顯示,衰老過程中的慢性應激會促進核RBPs FUS和PAB-1的出核,導致核RBPs在胞質中形成不溶性聚集體,而在通過減少IIS信號介導的長壽線蟲里HSF-1和DAF16被激活,從而消除這些蛋白質聚集體[83].這些都提示,在衰老過程中衰老細胞的內環境和代謝變化,會使得細胞監控系統受到干擾,從而可能影響SGs動力學,導致異常蛋白質聚集體易于形成.此外,與年齡性神經退行性疾病相關的RBPs突變同樣能引起SGs聚集和清除等動力學的異常[71,79,85],但RBPs的突變先引起SGs的動力學異常而導致疾病發生,還是先誘導疾病發生使SGs異常從而促進疾病發展,目前還沒有明確的定論,但無法否認的是,神經退行性疾病與異常SGs之間聯系緊密.
5 總結與展望
SGs是由LLPS驅動形成的一種壓力誘導的動態顆粒,由于其動態的蛋白互作網絡,SGs對環境變化非常敏感,使細胞能夠更靈活地應對這些變化,從而促進細胞在壓力下的生存.任何改變SGs關鍵組分的濃度、定位和結構構象的機制都將影響SGs的形成和動力學,導致其異常相分離[65-66].正常條件下,細胞受到應激形成SGs,并在應激撤去后解聚SGs,若形成異常SGs,則在PQC的調控下被靶向溶酶體或蛋白酶體清除.正常SGs通過隔離衰老調控因子延緩細胞衰老,而異常SGs由于無法被及時清除導致疾病發生.含有TDP-43,FUS或Ataxin-2的RBPs聚集是多種神經退行性疾病的關鍵特征,并且在其中發現一些SGs標記物,揭示了患者病理性RBPs聚集體的積累可能是SGs調節功能障礙所致[86-88,100].這些易于聚集的蛋白可以被招募到SGs中,起到成核作用,可能導致在細胞應激期間和應激消退后出現異常的SGs,難以解聚或被清除.而這些異常的SGs可能促進衰老細胞中不可逆的成熟蛋白質聚集體,加上細胞隨著年齡的增長抗壓能力降低,進一步加速這些蛋白細胞功能的衰退.因此,維持適當的SG動態可能是延緩衰老和延長壽命的一種潛在策略.
另一方面在年輕個體中,多種細胞防御系統可以保護細胞免受損害變化的影響,如蛋白質穩態紊亂和活性氧增加,因此,衰老細胞(至少是那些由急性應激激活p53誘導衰老的細胞)能被免疫系統有效地清除[101].然而隨著年齡的增長,一些衰老細胞獲得了逃脫清除的能力,隨著年齡的增加而逐年積累,不僅導致組織再生和修復的減少[5],還無法及時“修正”細胞內的異常,這種系統的年齡依賴性故障可能導致正常SGs組裝、解聚和清除方面的缺陷,這進而可能導致相關疾病的發生.目前比較確定的是,衰老細胞在急性應激中通過形成相對較多的SGs來響應應激,但無法清除已形成的SGs,極易形成不溶性聚集體導致衰老相關疾病發生;而暴露在慢性應激或持續性應激下的衰老細胞形成及清除SGs能力較弱,無法響應應激,導致細胞生存能力較弱.總而言之,衰老細胞的內環境惡化及功能退化抑制SGs形成,即使在急性應激下能誘導形成一定的SGs,也因為無法清除SGs而加劇甚至誘發疾病.目前還無法很好地解釋二者的差異,因此進一步的研究將有助于揭示SGs與衰老相互作用的機制,比如慢性應激與急性應激下,衰老細胞內環境變化引起的SGs組成成分差異,以及動態分析這些差異對SGs性能變化,而這些變化是否會產生協同效應,從而加速衰老.從這些研究中確定的關鍵成分可能產生新的檢測衰老標志物和有價值的藥物靶點,用于治療與衰老相關的疾病.另外,進一步了解幼齡和老齡動物SGs動力學調控的分子機制,同樣有助于識別對抗年齡相關性神經退行性疾病的治療新靶點.
猜你喜歡
鴨綠江(2021年35期)2021-04-19 12:24:18
考試與評價·高一版(2020年6期)2020-11-02 02:45:24
媽媽寶寶(2019年10期)2019-10-26 02:45:34
中國生殖健康(2019年3期)2019-02-01 06:12:26
鐵道通信信號(2018年11期)2019-01-19 01:15:08
電子制作(2018年11期)2018-08-04 03:25:42
鐵道通信信號(2018年2期)2018-04-18 12:18:10
鐵道通信信號(2016年11期)2016-06-01 12:11:32
鑿巖機械氣動工具(2016年3期)2016-03-01 04:00:25
中國病理生理雜志(2015年8期)2015-12-21 12:38:06
