分光光度法測定三唑錫可濕性粉劑中錫離子含量
2022-04-15 13:46:10姜宜飛熊安定吳進龍
農藥科學與管理 2022年2期
武 鵬,姜宜飛,李 蕊,熊安定,吳進龍*
(1. 農業農村部農藥檢定所,北京 100125;2.江蘇衡譜分析檢測技術有限公司,江蘇 南京 210046)
1 前言
三唑錫屬廣譜內吸性殺螨劑,其毒理機制與抑制神經細胞的氧化磷酸化過程有關,可以有效阻止體內ATP的形成;對敏感性的和具有抗藥性的螨類有很好的防治效果,可殺滅若螨、成螨和夏卵,對冬卵無效,殘效期>30d[1]。
2017年發布的《農藥登記資料要求》中規定農藥有效成分以鹽的形式存在時,需規定配對反離子的含量。目前國內已有三唑錫的分析方法,但有關三唑錫中錫離子的分析方法未見公開報道。本文采用分光光度法,對三唑錫中錫離子進行定量分析,該方法操作簡便、快速、準確,分離效果好,精密度和準確度均能達到定量分析的要求,可以作為企業生產過程質量控制和質檢機構分析檢測的參考方法。
2 試驗部分
2.1 試劑和溶液 乙醇(分析純);甲醇(分析純);酒石酸(分析純);抗壞血酸(分析純);酚酞(分析純);氨水(分析純);硫酸(分析純);硝酸(分析純);高氯酸(分析純);苯芴酮(分析純);水:新蒸二次蒸餾水或超純水;酒石酸溶液:ρ=100g/L;抗壞血酸溶液:ρ=10g/L(需現配現用);動物明膠溶液:ρ=5g/L(需現配現用);氨水溶液:Ψ(氨水∶水)=50:50;硫酸溶液:Ψ(硫酸∶水)=10:90;硝酸-高氯酸混合酸:Ψ(硝酸∶高氯酸)=80:20;苯芴酮溶液:稱取0.01g(精確至0.001g)苯芴酮,置于100mL容量瓶中,加入少量甲醇及數滴硫酸溶解,用甲醇稀釋至刻度,搖勻;酚酞指示劑:ρ=10g/L,按GB/T 603配制[2];25%三唑錫可濕性粉劑;金屬錫標樣:已知質量分數,ω≥98.0%。
2.2 儀器 分光光度計;控溫加熱板;2cm石英比色皿;25mL比色管。
2.3 測定步驟
2.3.1 錫標準儲備溶液的配制 稱取0.25g(精確到0.000 1g)金屬錫標樣,置于100mL燒杯中,加入10mL硫酸,蓋上表面皿,加熱至120 ℃至錫完全溶解,移去表面皿,繼續加熱至180 ℃產生濃白煙后,冷卻至室溫,緩慢加入50mL水,轉移至100mL容量瓶中,用硫酸溶液多次洗滌燒杯,將洗滌液并入容量瓶中,用硫酸溶液稀釋至刻度,搖勻備用[3]。
2.3.2 標準曲線的繪制 用移液管移取1mL錫標準儲備溶液,置于250mL容量瓶中,用硫酸溶液稀釋至刻度,搖勻;用吸量管移取0.1、0.2、0.4、0.6、0.8、1.0mL上述溶液,分別置于25mL比色管中,加入0.5mL酒石酸溶液和1滴酚酞指示劑,搖勻;加入適量氨水溶液中和至淡紅色,加入3mL硫酸溶液、1mL動物明膠溶液和2.5mL抗壞血酸溶液,用水稀釋至刻度,搖勻;加入2mL苯芴酮溶液,搖勻,靜置1h;同時制備空白溶液。以空白溶液為參比,于波長490nm處測定各標樣溶液的吸光度,以錫離子質量為橫坐標,吸光度為縱坐標繪制標準曲線。
2.3.3 試樣溶液的配制 稱取含0.005g(精確至0.000 1g)錫的試樣,置于50mL高腳燒杯中,加入10mL硝酸-高氯酸混合酸、2.5mL硫酸和3粒玻璃珠,混合均勻,蓋上表面皿;室溫通風放置24h后,加熱至120 ℃微沸,保持微沸消解4h,移去表面皿;繼續加熱至180 ℃消解,若消解過程中溶液過少或溶液變成黑色時,可加入適量硝酸;繼續消解至冒白煙,待溶液澄清,體積近2.5mL時,冷卻至室溫;將澄清液轉移至1 000mL容量瓶中,用75mL硫酸溶液分3次洗滌燒杯,每次超聲波振蕩5min;將洗滌液并入1 000mL容量瓶中,用水稀釋至刻度,搖勻。用移液管移取1mL上述溶液,置于25mL比色管中,加入0.5mL酒石酸溶液和1滴酚酞指示劑,搖勻;加入適量氨水溶液中和至淡紅色,加入3mL硫酸溶液、1mL動物明膠溶液和2.5mL抗壞血酸溶液,用水稀釋至刻度,搖勻;加入2mL苯芴酮溶液,搖勻,靜置1h;同時制備試樣空白溶液。以試樣空白溶液為參比,于波長490nm處測定試樣溶液的吸光度,在標準曲線上查得相應的錫離子質量。
2.3.4 計算 試樣中錫離子的質量分數ω(%),按式(1)計算:
(1)
式中:
ω——試樣中錫離子的質量分數,%;
m1——試樣溶液中錫離子的質量,μg;
V1——試樣消解液的定容體積(V1=1000),mL;
m2——試樣的質量,g;
V2——測定用試樣消解液的體積(V2=1),mL;
10 000——換算系數。
3 結果與討論
3.1 分析條件的選擇 本試驗方法主要參考 GB 5009.16-2014 《食品安全國家標準 食品中錫的測定》中第二種方法-苯芴酮比色法[3],試樣經消化后,在弱酸性溶液中四價錫離子與苯芴酮形成微溶性橙紅色絡合物,在保護性膠體存在下與標準系列溶液比較定量。但本試驗中三唑錫可濕性粉劑的組份組成比較復雜,干擾物較多,試樣溶液制備時采用國標中硫酸消解的試驗效果不理想;采用硝酸時,硝酸雖能破壞絕大部分有機物,但易在試樣完全溶解前沸騰,且溫度較高時容易分解;采用硝酸-高氯酸混合酸直接高溫消解易發生爆炸,所以本方法采用10mL硝酸-高氯酸混合酸、2.5mL硫酸混勻后進行消解。與國標方法相比,酸與試樣混勻后的放置時間和加熱消解時間都進行了延長調整,保證三唑錫可濕性粉劑中錫的消解完全,通過試驗摸索將消解條件調整為:將酸與試樣混勻后室溫通風放置24h,然后加熱至120 ℃微沸消解4h,然后將消解溫度提高至180 ℃,直至冒白煙,消解液變澄清時,表明試樣完成消解。經試驗驗證標樣采用國標和本試驗的消解方法對試驗數據沒有顯著影響,基于安全和簡便考慮,本試驗中標樣的消解方法仍采用國標中的消解方法。
3.2 分析方法的線性相關性試驗 按照2.3.2繪制校正曲線。試驗測得錫離子線性方程為y = 0.021 5x + 0.003 3,相關系數為0.999 2。結果表明三唑錫中錫離子在測試的質量范圍內線性關系良好。
3.3 分析方法的精密度試驗 準確稱取5個試樣,按照2.3測定步驟進行試驗分析,測得三唑錫試樣中錫離子的標準偏差為0.06,變異系數為0.84%(表1)。

表1 分析方法的精密度試驗結果
3.4 分析方法的準確度試驗 稱取5個已知錫離子質量分數(7.13%)的試樣,分別加入一定量的錫標準儲備溶液,按照2.3測定步驟進行分析,測得三唑錫試樣中錫離子的平均回收率為97.9%(表2)。
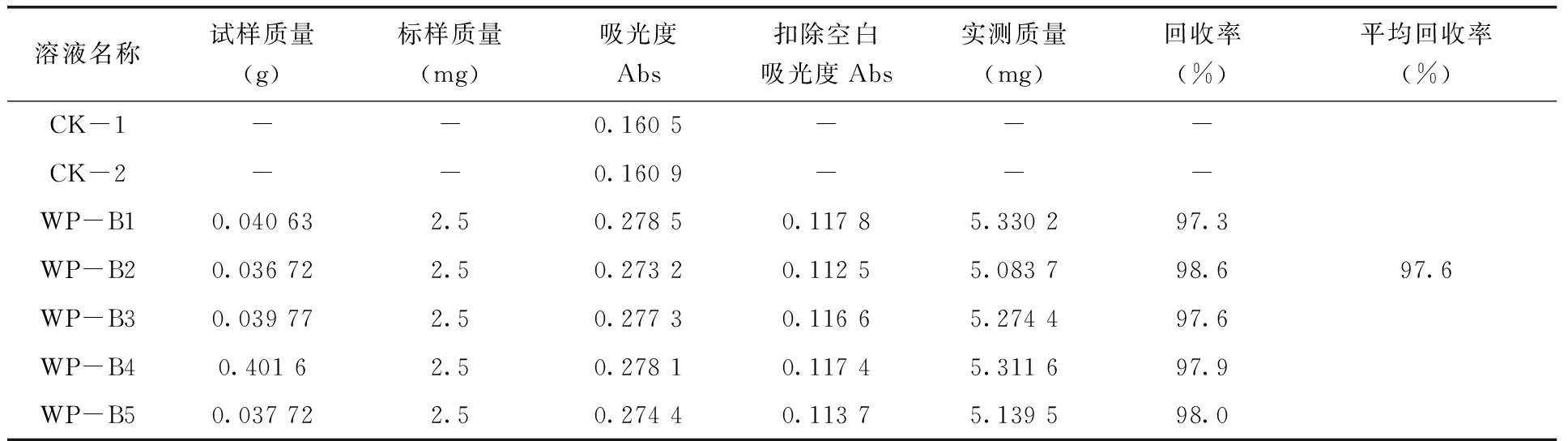
表2 分析方法的準確度試驗結果
4 結論
試驗建立了分光光度法測定三唑錫中錫離子質量分數的分析方法。試驗結果表明,錫離子在測試質量范圍內線性關系良好,方法精密度和準確度較高,具有操作簡便、快速的特點,是產品質量控制和應用研究中較為理想的分析方法。
