固態核磁共振技術在晶型藥物研究中的應用*
2022-04-27 03:08:58陳婷李琦劉琪文楊世穎張麗呂揚
醫藥導報 2022年5期
關鍵詞:信息
陳婷,李琦,劉琪文,楊世穎,張麗,呂揚
(北京協和醫學院、中國醫學科學院藥物研究所晶型藥物研究北京市重點實驗室,北京 100050)
根據藥物內在結構,固態藥物可分為晶態物質和非晶態物質,具體可以包括多晶型、共晶、鹽、溶劑合物、水合物及無定型等多種存在形式。藥物分子在固態下不同排列形式通常會引起溶解度、穩定性、生物利用度和吸濕性等的巨大變化[1]。藥物存在2種或2種以上的不同晶型物質狀態,被稱為存在多晶型現象。多晶型現象不僅影響藥物的理化性質,對其藥理活性等也會產生很大影響,同種藥物的不同晶型在臨床治療效果上可能有很大差異[2]。藥物共晶是調節活性藥物成分(active pharmaceutical ingredient,API)性質的有效方法,通過選擇合適的共晶形成物(cocrystal former,CCF),發生特定的非共價相互作用,從而改變API的溶解性、穩定性、滲透性、生物利用度等重要性質[3]。無定型狀態是一種特殊的物質晶型狀態,無定型狀態藥物的各種理化性質及臨床藥效特征常有別于一般的晶型藥物。其分子處于熱力學不穩定狀態,具有更高的自由能,能在一定程度上改善API的溶解速度與程度[4]。
20世紀70年代以來,固態核磁共振(solid-state nuclear magnetic resonance spectroscopy,SS-NMR)技術在藥物分析領域發揮著越來越重要的作用[5]。SS-NMR能夠提供原子水平的結構信息,且通常以化學位移的形式反映原子核的局部電子環境和核間距情況[6]。SS-NMR對分子內晶體堆積效應及弱分子內非共價鍵非常敏感,在識別化合物及確定分子結構和獲得動力學參數方面具有重要意義[7]。筆者概述SS-NMR的技術進展及在晶型藥物研究中的應用,為使用該技術方法更充分更全面地研究晶型藥物提供參考。
1 SS-NMR進展
1.1SS-NMR法概況 SS-NMR可以提供固態藥物的很多信息,如短程無序的狀態、氫鍵性質、鹽或共晶特征、水或溶劑分子的存在、晶胞中獨立分子的數量(Z′)和晶體不對稱單元中分子的數量(Z0)等[8]。SS-NMR中常見的核磁共振相互作用包括:①磁屏蔽效應,這導致了化學位移張量。化學位移提供了許多固態結構和晶體堆積相互作用的信息,化學位移分配是從核磁共振譜中提取結構和動力學信息的先決條件[9];②偶極耦合(直接耦合),偶極耦合來源于相鄰核自旋間產生的磁場的相互作用,通過測量一對原子核之間的偶極耦合通常能夠獲得原子間距離等信息[10];③J耦合(間接耦合),與偶極耦合相反,J耦合相互作用是由電子介導的,可通過計算電子結構獲得J耦合相關信息[11];④核電四極耦合,四極相互作用會使圖譜線條變寬,對其進行分析可以獲取原子核局部環境的信息[12]。利用SS-NMR分析藥物的主要參數有化學位移,偶極相互作用,化學位移各向異性,弛豫時間(T1)和同位素效應等[8]。合理運用這些參數對獲取藥物結構內部信息具有巨大幫助。
1.2SS-NMR常用技術 近年來,SS-NMR的分辨率和靈敏度得到了很大的提高,增強了表征固體藥物的能力[13]。相關技術的進步極大地提高了獲取高分辨率光譜的能力,從而能夠獲取原子核局部電子環境和核間距的豐富信息。
60 kHz或更高轉速下的超快魔角旋轉技術(ultrafast magic angle spinning technology,UF-MAS)極大提高了SS-NMR的分辨率和靈敏度,如圖1所示[14],目前,轉速超過100 kHz的魔角旋轉探頭已經成功運用[15]。MAS是指將樣品繞著相對于外部磁場B0傾斜54.736°的軸進行物理旋轉,利用此方法可以達到消除各向異性、偶極耦合和J耦合的效果[11]。1H-14N 相位調制旋轉回波飽和脈沖雙共振序列(phase-modulated rotational-echo saturation-pulse double-resonance sequence,PM-RESPDOR)作為一個新的序列,可在非常高的轉速下通過1H MAS檢測來確定1H-14N的距離[16]。在高速魔角旋轉條件下,對1H化學位移的高靈敏度可用來區分多晶型物,DAMRON等[17]通過SS-NMR區分了無法利用振動光譜法明確區分的對乙酰氨基酚多晶型物。
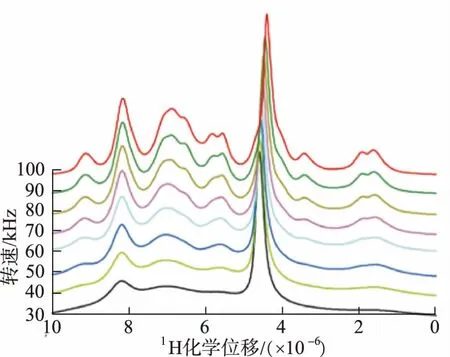
圖1 30~100 kHz的轉速下獲得的1H MAS NMR譜圖Fig.1 1H MAS NMR spectra obtained at spinning frequencies varying from 30 to 100 kHz
交叉極化(cross polarization,CP)在固態核磁領域也是最常用的方法之一,其特殊激發途徑不僅可以提供更高的信號強度(低天然豐度的原子核),并且也提高了采集效率[11]。一維(1D)的CP實驗是常用的質譜核磁共振技術,可提供了關于分子結構、動力學和藥物相互作用的豐富信息[13]。CP與MAS相結合的方法已成為提高SS-NMR檢測靈敏度的重要手段[18]。伴隨技術的發展,多維、多核SS-NMR實驗逐漸克服了需要大量樣品的缺點。一維13C CP/MAS和二維1H-13C 異核相關性實驗,不僅顯著提高了信噪比,并且證明了利用少量樣品(約2.0 mg)進行高分辨率13C MAS實驗的可能性[19]。
動態核極化(dynamic nuclear polarization,DNP)是一種電子-核的雙共振極化技術,用微波激發自由電子躍遷,使相關核的自旋能級分布發生極化來增強靈敏度[20]。近年來由于高場DNP技術的發展,NMR的檢測靈敏度提高了量級2~3個[21]。DNP的高靈敏度有助于快速獲取13C和15N固態核磁共振譜,實現2D1H-13C、1H -15N固態核磁共振實驗,在多晶型鑒別、核磁共振晶體學結構測定以及探測各種有機材料中的氫鍵方面發揮著重要作用[22]。
1.3常用的固體核磁共振探針 藥物中包含的核磁共振敏感核不僅有最常見的1H和13C,還有19F、14N、15N、15O、23Na和31P等[23](表1[9,21])。自旋量子數I=1/2的原子核(1H、13C、15N等),可以通過化學屏蔽效應(即周圍電子對局部磁場的改變)和核間耦合來獲得信息[11]。因此,通過分析原子分辨率可以獲得關于原子之間連接方式以及單個原子周圍化學環境的重要信息[24]。
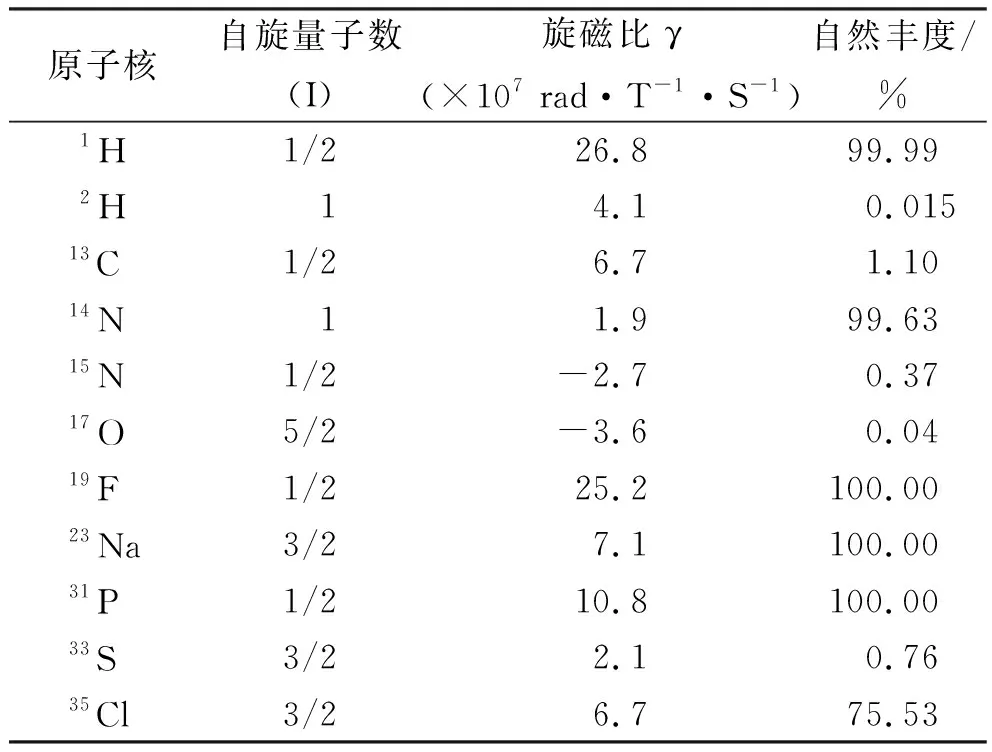
表1 藥物中包含核磁共振敏感核的信息 Tab.1 Information on NMR-sensitive nuclei contained in drugs
針對某一特定原子核的測定,可以確定正在研究的元素;在多核測量的情況下還能獲取更多的結構信息。13C NMR是平均各向異性相互作用的有效手段,可以用來探測氫鍵的破壞和形成[25]。針對13C NMR開發的濾波器-選擇性信號激發方法,可從藥物中有目的地獲得13C信號。另外13C NMR在固態藥物定量分析方面的應用也極為成熟[26]。
除藥物研究中廣泛使用的13C自旋核,1H也是區分結晶形式的常用核。1H自旋核位于分子表面,質子共振對晶體堆積非常敏感,所以可獲得比13C NMR更高的分辨率、靈敏度和采集速度[27]。1H NMR是探測氫鍵是否存在的強有力手段[28],對于解析晶體結構內部信息有很大的幫助。
從1H和13C天然豐度水平的自旋核中容易獲得核磁共振信號,所以大多數藥物分子的SS-NMR研究都使用1H或13C作為核磁共振探針。但是也有研究表明,使用17O NMR獲得的SS-NMR圖譜可以補充1H和13C核磁共振圖譜數據[29]。另外,因為氫鍵會影響17O核磁共振參數,所以17O NMR也可以探測氫鍵的形成位點[30]。
氮元素普遍存在于胺、酰胺等官能團中,以及氮在固體原料藥中可能形成分子間氫鍵,天然存在的具有核磁共振活性的氮核14N 和15N在解析結構時可以發揮重要作用[31]。因為15N具有良好的I=1/2特性和相對于14N更高的旋磁比,藥物的氮SS-NMR研究主要集中在15N上。但由于15N天然豐度較低(0.37%),實驗耗時長,而14N天然豐度高(99.6%),不需要同位素標記,14N NMR正得到越來越多的應用。另外,14N四極相互作用通過電場梯度參數能夠提供15N-NMR核磁共振研究無法獲得的結構信息[32]。
13C和15N化學位移更好地揭示了共晶體或鹽的形成,1H、13C和15N等核磁共振化學位移數據可用于檢測和估計氫鍵的存在及強度。因此,基于不同核的優勢選擇單核或多核測量使SS-NMR可用于鑒定原料藥的多晶型物、共晶及溶劑化物等,提供原料藥的詳細結構信息。
1.4SS-NMR與計算方法相結合 無論何種光譜,提取準確的NMR參數并分析其中包含的結構信息仍然是一個挑戰。只有在相關計算方法的幫助下,NMR光譜學提供的許多結構信息才能得到充分利用[11]。
VENANCIO等[33]利用1H MAS NMR技術與第一原理計算測量投影綴加波(gauge-including projector-augmented wave,GIPAW)相結合的方法探測到枸櫞酸乙胺嗪鹽的分子間相互作用,促進了核磁共振技術在表征藥物共晶領域中的應用。BLADE等[34]基于密度泛函理論(density functional theory,DFT)的GIPAW計算方法,結合溶液和SS-NMR實驗數據,成功地區分了托芬那酸的4種多晶型物。在量子力學的幫助下,SS-NMR能夠更好地解析晶體結構,包括多晶型檢測和表征、檢測溶劑化物的存在、研究無定型等[21]。從頭隨機結構搜索(ab initio random structure searching,AIRSS)是一種基于DFT的晶體結構預測方法,MATHEW等[35]采用AIRSS并聯合SS-NMR所提供的化學位移值區分了阿司匹林結構相似的晶Ⅰ型和Ⅱ型。涉及晶體結構預測的核磁共振結晶學(nuclear magnetic resonance crystallography,NMRX)方法也已經用于確定粉末的晶體結構[36]。
將多種方法結合起來,能夠達到比任何一種單獨的方法更強的洞察力。未來寄希望于推動SS-NMR實驗與其他不同的計算方法相結合,從而增強實驗數據的準確性與提高SS-NMR的洞察力。
1.5SS-NMR與其他表征手段聯用與比較 固態藥物分析傳統技術有單晶X射線衍射法(single crystal X-ray diffraction,SXRD)、粉末X射線衍射法(powder X-ray diffraction,PXRD)、差示掃描量熱法(differential scanning calorimetry,DSC)、熱重分析法(thermogravimetric analysis,TG)、紅外光譜法(infrared spectroscopy,IR)、拉曼光譜法(Raman spectroscopy)等。
與液態核磁共振技術(liquid-state NMR,LS-NMR)不同,SS-NMR分析的樣品為粉末狀固體,因此可以直接提供樣品在固態下的特征信息,可用于獲取液體狀態下消失或轉變的藥物結構信息。另外,LS-NMR在表征大多數有機固體時,通常會產生一個寬的、無特征的譜圖[6]。相比之下,CP和MAS等技術的發展極大地緩解了SS-NMR譜線增寬問題。
SXRD是確定藥物晶體結構的國際權威方法。但是該方法也受到一定限制,即需要有適合單晶X射線衍射實驗用的單晶體。衍射實驗對單晶體尺寸有一定的要求(尺寸≥0.1 mm),因此培養單晶常常成為耗時、耗力的瓶頸環節。此外,為了獲得晶體工程中特別重要的非共價相互作用的正確信息(如氫鍵),需要明確氫原子的位置[37],而SXRD有時不能提供精確的氫原子位置,也無法探測主體材料中客體分子的運動和無序的情況[12,38]。SS-NMR通常能彌補SXRD的這些缺點,對于探測短程有序而長程無序的固體(如無定型)具有重要意義[39]。鑒于衍射方法和SS-NMR對長程有序和短程環境的靈敏度具有天然的互補性,二者聯用可以獲得更全面的結構信息。
PXRD是表征晶型藥物強有力的技術,在已知晶體結構,SS-NMR與PXRD結合能提供比其中單一技術更詳細的信息。在基于DFT的GIPAW計算方法的幫助下,13C SS-NMR實驗數據進一步驗證了由PXRD確定的鹽酸西咪替丁的晶體結構[40]。
紅外光譜與拉曼光譜聯用能相互佐證,提供相對全面的結構信息。與拉曼光譜和紅外光譜不同,核磁共振譜中的峰面積能提供官能團的相對濃度[9]。熱分析技術在晶型藥物的研究中具有不可替代的作用,其中最常用的TG和DSC是粉末表征的有效且準確的方法,二者聯用通常可以分析熱力學差異和計算溶劑的種類和數量[41]。
在晶型藥物研究中,基于SS-NMR與其他表征手段如衍射法的優勢互補,SS-NMR與其他技術的聯用具有某一單一技術所沒有的優勢,可使獲得的信息更全面以及更具說服力。
2 SS-NMR在藥物多晶型中的應用
2.1藥物多晶型 原料藥的多晶型現象是由于分子的不同堆積方式、分子構型構象差異、結晶水和結晶溶劑介入或分子作用力差異等原因引起的。據估計,超過50%的API存在多晶型現象[42]。對藥物多晶型的深入研究,首先為最大限度地降低長期儲存時相變和產品性能變化的風險,開發臨床有效的優勢藥用晶型[43]。其次,一種新的藥物晶型可以延長專利時間,為原研企業創造最大程度的利益,同時也能讓仿制藥公司在不侵犯原研專利的情況下生產其產品[44]。深入SS-NMR在藥物多晶型領域中的研究,對突破API臨床應用的限制很重要。
2.2在藥物多晶型領域的定性分析應用進展 在藥物多晶型的研究中,其中重要的一項是區分藥物晶型種類,SS-NMR可用于鑒定和鑒別API多晶型物。在估計分子構象的數量以及晶胞中分子堆積的差異方面,13C SS-NMR 具有顯著的優勢[45]。自旋晶格弛豫時間T1取決于樣品中的分子運動,可探測拉莫爾頻率下磁漲落的譜密度,并隨著溫度的降低而增加。APIH等[46]用核磁共振氫譜研究了卡馬西平的多晶型物。4種晶型的質子在室溫和VL=32 MHz條件下的T1從晶Ⅳ型的0.40 s、晶Ⅱ型的0.52 s、晶Ⅳ型→晶Ⅲ型的50 s、晶Ⅲ型的100 s到晶Ⅰ型的195 s不等。當樣品冷卻到165 K時,晶Ⅳ型的T1增加到30 s,而Ⅰ、Ⅱ和Ⅲ型的T1分別減少為100,0.45和55 s。在不同溫度下測量質子的T1從而達到區分API不同多晶型的目的。另外,SS-NMR還可以作為PXRD結構分析的補充手段。除T1外,化學位移值是區分多晶型物的重要參數。利用API中的氯元素,NAMESPETRA等[47]成功使用35Cl MAS NMR表征鹽酸異丙腎上腺素和美西律形成的鹽酸鹽多晶型物。最新開發的一種光學過程采用了基于DFT-SS-NMR計算和實驗固態核磁共振數據之間差異分布的化學位移計算和貝葉斯概率論。在實驗數據非常相似的情況下,如當XRPD或13C核磁共振產生不明確的結果時,將DFT方法應用于13C核磁共振化學位移預測并結合概率論,能精確識別多晶型物[48]。
SS-NMR在研究溶劑合物及水合物形成方面具有獨特優勢。利用CP/MAS SS-NMR可以區分茶堿的2種無水物(晶Ⅰ型、晶Ⅱ型)及一水合物。1H和13C CP/MAS SS-NMR的譜圖顯示,3種多晶型中胺和甲基質子的化學位移發生變化。在晶Ⅱ型中NH中的質子與氮原子形成氫鍵,晶Ⅰ型兩個茶堿分子中,NH基團與相鄰茶堿分子的羰基形成氫鍵,光譜如圖2所示[49],3種多晶型物之間化學位移的差異可作為區分其晶型的重要特征。
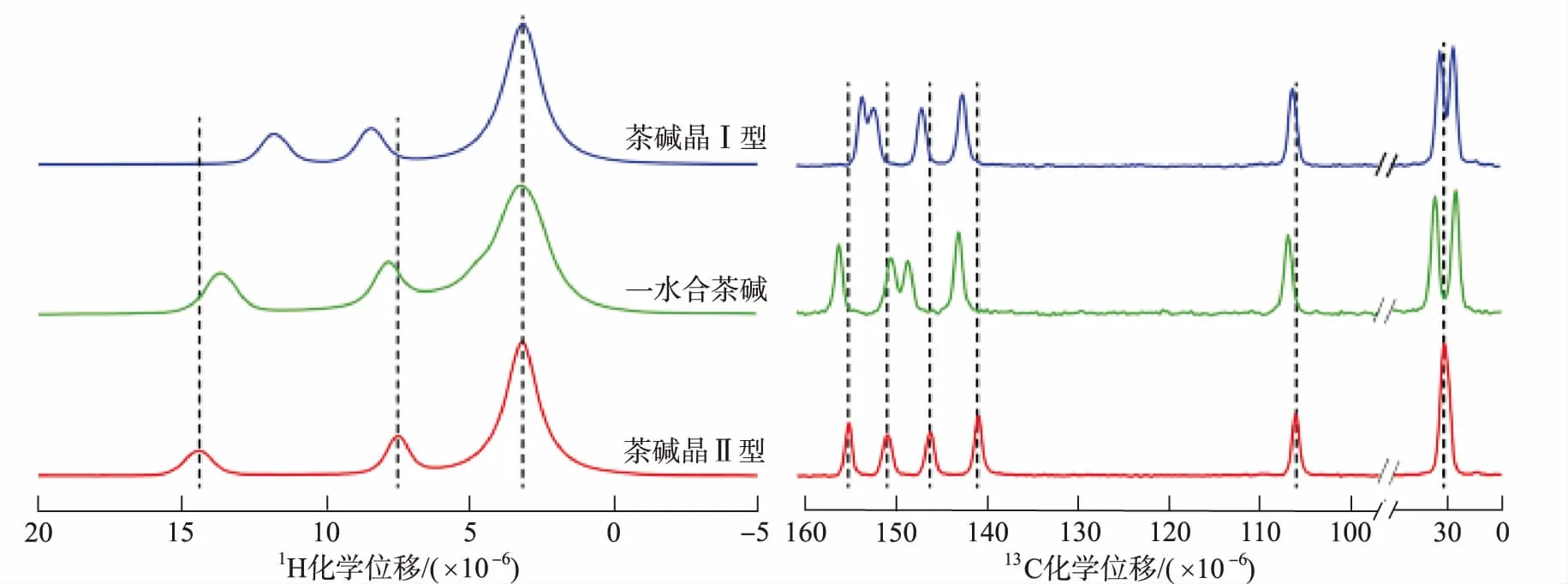
圖2 茶堿晶Ⅰ型(theo-Ⅰ)、一水合茶堿(theo-m)和茶堿晶Ⅱ型(theo-Ⅱ)1H和13C CP/MAS SS-NMR譜圖 Fig.2 1H and 13C CP/MAS SS-NMR spectra of theophylline form Ⅰ (theo-Ⅰ),theophylline monohydrate (theo-m),and theophylline form Ⅱ (theo-Ⅱ)
SS-NMR在分析API的多晶型相變,以及固體制劑分析中的多晶型鑒定方面也發揮著重要作用。吡拉西坦多晶型物的相對穩定性依賴于溫度,SZELESZCZUK等[50]利用SS-NMR結合DFT計算方法,發現DFT計算不僅可以得到核磁共振參數用于分析多晶型相變,還可以預測溫度對多晶型物穩定性的影響。
2.3在藥物多晶型領域的定量分析應用進展 磁等效位置的信號強度與相應的自旋數成正比,因此核磁共振測量可以提供樣品成分含量的準確信息,可用于定量測定[51]。13C CP/MAS SS-NMR是一種精確的技術,在一定條件下樣品中的碳元素與該相的濃度成比例時,可以達到量化聚合物的目的。因此,CP/MAS SS-NMR可獨自作為定量測定原料藥和藥品中相純度或者比例的方法[52]。
SS-NMR能量化并精確地檢測藥物多晶型的存在。RODNEY等[53]通過常規的13C CP/MAS SS-NMR,利用藥物多晶型的核磁共振譜的差異來分析不同晶型的物理混合物,建立校準曲線用于羅昔非班晶Ⅰ和Ⅱ型的定量。SS-NMR在藥物表征中的主要任務是定量分析API在藥物和固體制劑中的晶型變化。結合13C SS-NMR和PXRD圖譜的信號證明了在賦形劑存在的情況下,多晶型物的可重復和準確的定量[54]。厄貝沙坦是Avalide片劑中的活性藥物成分,有2種晶型即晶A型和晶B型。厄貝沙坦晶A型是Avalide片劑濕法制粒中優勢藥用晶型,B型溶出度較低。利用近紅外光譜和SS-NMR定量檢測阿瓦利德片中極少量的厄貝沙坦晶B型,確定了原料藥的晶型純度[55],對于藥品質量控制做出了極大貢獻。
3 SS-NMR在藥物共晶領域的應用
3.1藥物共晶 40%的市售藥物和高達90%的新化學實體都表現出較差的水溶性,屬于生物制劑學分類系統的Ⅱ類和Ⅳ類,生物利用度較低[56]。API與CCF通常通過氫鍵、π-π堆積和范德華力等非共價相互作用而結合,可在不改變API內在結構的基礎上改善其理化性質,如溶解度、吸濕性、藥理活性等[57]。SS-NMR在一定程度上可獨自探究藥物共晶的相關特性,也可作為X射線衍射和熱分析等方法的有效補充,在藥物共晶領域發揮重要作用。
3.2在藥物共晶領域的定性分析應用進展 SS-NMR通常能夠檢測共晶氫鍵形成的具體位置,這對于解析共晶的結構很必要。二維1H-14N HMQC SS-NMR在探究氫鍵形成位點方面具有很大的潛力,在14N-1H HMQC光譜中觀察到的N1-H1相關峰,表明煙酰胺棕櫚酸共晶形成分子間氫鍵,通過分析分子內NH交叉峰,確定了共晶的氫鍵位置為N1…H1-O2[28]。ROSSI等[58]通過SS-NMR、PXRD和基于DFT的實驗和計算相結合的方法對茶堿-鹽酸吡哆醇共晶進行了表征,確定了茶堿-鹽酸吡哆醇共晶的形成。譜圖3顯示13C化學位移發生在C2'=O12'的羰基部分,其共振從149.8×10-6(THEO原料藥)變為152.7×10-6(THEO·PyrH+Cl-),揭示2個分子片段之間形成O11-H11…O12'及弱C8′-H8′…O12′氫鍵聯系(O11…O12′和C8′…O12′距離分別為2.703(5) ?和3.593(4) ?。THEO·PyrH+Cl-的15N CP/MAS光譜(圖4)中THEO·PyrH+Cl-信號向高頻方向移動到231.1×10-6(Δδ=13.3×10-6),證明N9′沒有參與任何相互作用,說明H10與O11形成分子內O10-H10…O11氫鍵而不是O10-H10…N9′分子間氫鍵。13C SS-NMR在評估相純度和共晶形成程度方面對PXRD起到有價值的補充作用。MASHHADI等[59]聯合使用SS-NMR、PXRD、DSC對異煙肼-肉桂酸共晶進行了表征,并用SXRD確定了該共晶的結構。通過觀察個別的13C化學位移,可以區分不同的氫鍵排列,為SXRD分析結構提供了有價值的補充和佐證。
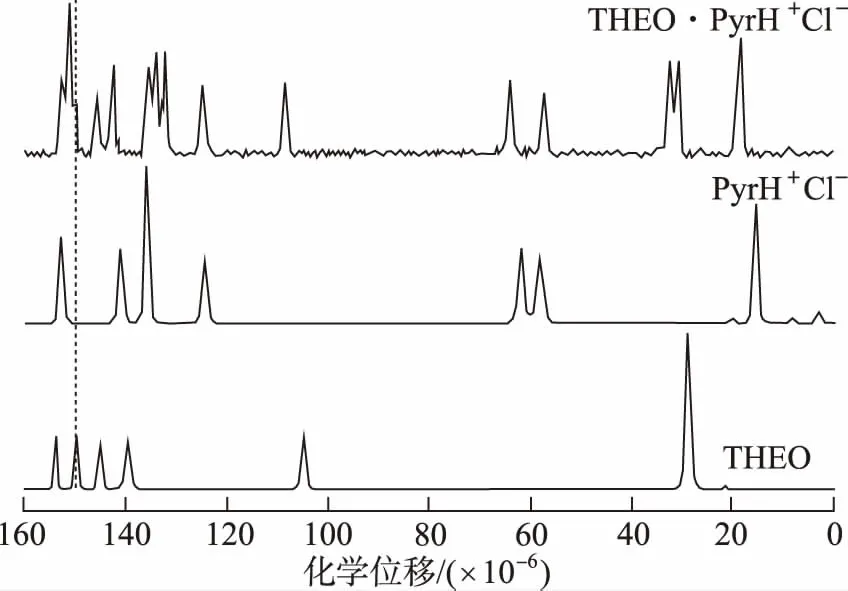
圖3 THEO、PyrH+Cl-、THEO·PyrH+Cl-的13C CP/MAS NMR譜圖 Fig.3 13C CP/MAS NMR spectra of THEO,PyrH+Cl- and THEO· PyrH+Cl-
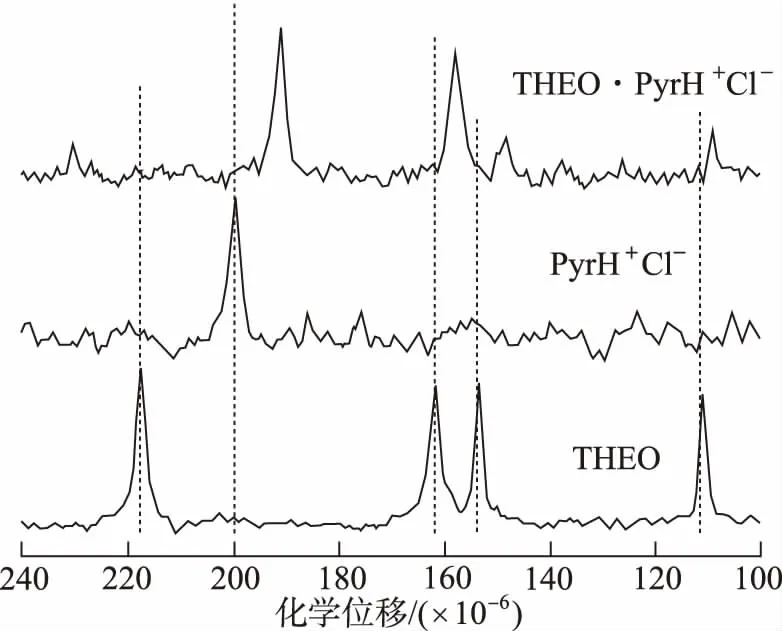
圖4 THEO、PyrH+Cl-、THEO·PyrH+Cl-的15N CP/MAS NMR譜圖 Fig.4 15N CP/MAS NMR spectra of THEO,PyrH+Cl- and THEO·PyrH+Cl-
確定CCF和API之間是否發生酸堿反應而形成鹽是藥物共晶領域的一個關鍵問題[60]。鹽存在質子的轉移,但對于共晶而言,API和CCF保持中性[61]。徐巾超等[62]通過SS-NMR得到的羅司他韋4個共晶的譜圖,結果顯示所形成的共晶均沒有發生質子轉移,證明了形成物是共晶而非鹽。
4 SS-NMR在藥物無定型中的應用
4.1無定型與共無定型 提高難溶性藥物的口服生物利用度是當前藥物開發最具挑戰性的方面之一。藥物無定型因熱力學不穩定而增加藥物的表觀溶解度和溶解速率[4],因此API的無定型制劑可以成為解決其低溶解度問題的一種替代方法。
藥物共無定型是指兩個或多個分子相互作用形成同質非晶態單相體系,基于組分間分子相互作用,如氫鍵或π-π作用等。除了提高溶解性,共無定型還能防止在儲存時重結晶[63]。與PXRD和熱分析技術相比,SS-NMR具有探測無定型分子堆積的能力,可獲得無定型局部結構信息。
4.2在藥物無定型領域的應用 一般而言,很難通過衍射技術對藥物無定型進行深入研究,以SS-NMR為主要研究工具,輔助以其他表征手段,可以獲取無定型內部結構和分子遷移率等重要信息,提高對藥物無定型的洞察力[64]。13C SS-NMR可檢測和量化無定型氫鍵相互作用程度,YUAN等[65]通過13C SS-NMR實驗,表明無定型中59%的吲哚美辛分子通過羧酸環狀二聚體氫鍵結合,15%在無序羧酸鏈中,19%通過羧酸和酰胺氫鍵結合,其余7%不含氫鍵。13C CP/MAS SS-NMR可探測β-氮雜萘啶-馬來酸共無定型中C-H…O氫鍵[66],推動了SS-NMR在共無定型結構表征中的應用。
5 結束語
無論是對基于改善藥物成藥性的新藥研發,還是基于療效與質量的仿制藥一致性評價,藥物的晶型研究都十分必要。因此,對晶型藥物進行全面深入的表征分析是認識其內在本質的關鍵。SS-NMR作為一種強有力的分析手段,具有定性鑒別、定量分析、探測藥物內部結構、分析多晶型相變等多種功能,這對于制劑中晶型藥物雜質的控制和含量測定無疑具有巨大幫助。SS-NMR的進步極大提高了檢測分辨率和靈敏度,克服了樣品微量和無定型性質低靈敏度的限制,使藥物多晶型、藥物共晶及無定型的表征分析更加全面、深入。需要注意的是,任何一種表征手段都有其固有的局限性,晶型藥物的研究與表征需聯合使用多種分析手段從不同角度出發,相互補充和完善才更可靠。在基于計算的基礎上,加強SS-NMR與衍射技術、熱分析方法等其他多種表征手段的聯用,無疑將促進對晶型藥物本質的認識,也將推動我國晶型藥物研究的發展。
猜你喜歡
中華手工(2017年2期)2017-06-06 23:00:31
中外會展(2014年4期)2014-11-27 07:46:46
大眾創業(2009年10期)2009-10-08 04:52:00
數字社區&智能家居(2009年7期)2009-09-29 08:16:48
數字社區&智能家居(2009年11期)2009-06-25 04:30:34
數字社區&智能家居(2009年3期)2009-04-21 03:09:04
數字社區&智能家居(2009年2期)2009-03-27 04:33:44
數字社區&智能家居(2009年12期)2009-02-03 07:50:48
建筑創作(2001年3期)2001-08-22 18:48:14
祝您健康(1987年3期)1987-12-30 09:52:32
