基于生物信息學(xué)的肥厚型心肌病易感基因TNNC1單核苷酸多態(tài)性致病表型分析
2022-05-18 07:23:52黎江溪張世梅王玉鑫
中國生物醫(yī)學(xué)工程學(xué)報 2022年1期
關(guān)鍵詞:數(shù)據(jù)庫分析
黎江溪 張世梅 王玉鑫 趙 躍
(大理大學(xué)基礎(chǔ)醫(yī)學(xué)院,云南大理 671000)
引言
肥厚型心肌病(hypertrophic cardiomyopathy,HCM)是一種常見且復(fù)雜的遺傳性心臟疾病,常引發(fā)青少年及年輕運動員的心源性猝死[1]。正常心肌細(xì)胞規(guī)則地組裝成平行排列的肌纖維,但HCM患者的肌纖維短寬肥厚,彌漫性肥大,排列紊亂,會引起舒張期功能障礙。眾多證據(jù)表明,突變的心臟肌節(jié)蛋白能增加肌絲對Ca2+的誘捕,導(dǎo)致Ca2+循環(huán)紊亂[2],其結(jié)果是產(chǎn)生代償性的心肌細(xì)胞肥大。
肌鈣蛋白位于肌動蛋白絲上,是橫紋肌收縮的中央調(diào)節(jié)蛋白,包括肌鈣蛋白T2、肌鈣蛋白I3 及肌鈣蛋白C1(cardiac troponin C1,TNNC1)3 個亞基[3]。TNNC1 作為編碼與Ca2+誘捕敏感蛋白的最重要基因之一,代表了心臟肌小節(jié)的Ca2+敏感元件。因此,TNNC1 基因結(jié)構(gòu)和功能是否正常,決定著粗細(xì)肌絲能否正常滑行[4]。有關(guān)TNNC1 基因突變與HCM 發(fā)病的分子遺傳學(xué)研究較少,在dbSNP 數(shù)據(jù)庫中僅發(fā)現(xiàn)A8V、A31S、E134D、C84Y 和D145E 與HCM 發(fā)病相關(guān)[5-6],該基因尚存在的其他非同義單核苷酸多態(tài)性(non-synonymous single nucleotide polymorphisms,nsSNPs)與HCM 疾病表型的關(guān)系還有待挖掘。因此,本研究利用生物信息學(xué)方法,對dbSNP 數(shù)據(jù)庫中TNNC1 基因的1 559 個SNPs 位點進(jìn)行整合分析,篩選出18 個與疾病關(guān)聯(lián)的有害nsSNPs。該研究以TNNC1 基因為示范,分析了其nsSNPs 與疾病表型的關(guān)系,這對HCM 的基因突變篩查、輔助臨床診斷及有效藥物的研發(fā),有著十分重要的理論指導(dǎo)意義。
1 材料和方法
1.1 基因數(shù)據(jù)收集和篩選
TNNC1 的基因序列(NG_008963.1)、蛋白質(zhì)序列(NP_003271.1)及SNP 位點等信息,均來自于美國國立生物信息中心dbSNP 數(shù)據(jù)庫(http:/ /www.ncbi.nlm.nih.gov/)。在分級篩選過程中,把TNNC1基因5′和3′UTR、內(nèi)含子及同義突變的SNP 去除,即可得到nsSNPs。
1.2 基因突變位點有害性分析
基于Mutation Taster 和PolyPhen-2 在線軟件,對初步篩選獲得的nsSNPs 進(jìn)行有害性分析。Mutation Taster 軟件整合了較為權(quán)威的外顯子組和千人基因組計劃數(shù)據(jù)庫,主要是根據(jù)氨基酸替代矩陣,兼顧氨基酸的物理化學(xué)特征以及氨基酸的差異程度,利用貝葉斯分類算法來預(yù)測突變造成的潛在疾病[7]。結(jié)果可定性描述為 disease causing (致病的)和Polymorphism(多態(tài)性)。預(yù)測評分可在0.00~215,評分越高,代表可信程度越高,當(dāng)評分>90,認(rèn)為是可信的。PolyPhen-2 軟件則是根據(jù)突變氨基酸序列的結(jié)構(gòu)和功能等方面,對被替換的氨基酸進(jìn)行分析[8],預(yù)測得分范圍在0.00~1.00。當(dāng)評分為0.00~0.49,預(yù)測為良性(benign);0.50 ~0.89 分,預(yù)測為可能損傷(possible damaging),0.90~1.00 分,預(yù)測為很可能損傷(probably damaging)。
在Mutation Taster 及PolyPhen-2 軟件有害性分析的基礎(chǔ)上,運用PhD-SNP 及MutPred 軟件,對nsSNPs 與疾病相關(guān)性做更深入的分析。PhD-SNP軟件基于SVM 算法,主要對目標(biāo)nsSNPs 中對應(yīng)的氨基酸突變位置及突變前后的數(shù)據(jù)集進(jìn)行疾病關(guān)聯(lián)分析,當(dāng)評分≥0.4 時認(rèn)為可信度較高,預(yù)測結(jié)果判定為中性(neutral)或疾病(disease)[9]。MutPred是基于機器學(xué)習(xí)的軟件,通過可能影響表型的特定分子變化排序列表來集成分子數(shù)據(jù),最后確定氨基酸取代后可能的致病性。結(jié)果可描述為假設(shè)非常可信、假設(shè)可信和假設(shè)可操作[10]。
1.3 蛋白三維模型構(gòu)建及分析
基于Swiss Model 數(shù)據(jù)庫(https:/ /swissmodel.expasy.org/interactive/),對前期篩選到的與疾病關(guān)聯(lián)的nsSNPs 進(jìn)行三維結(jié)構(gòu)模型構(gòu)建。最后利用VMD 軟件[11],對突變前后蛋白結(jié)構(gòu)模型進(jìn)行可視化分析。
2 結(jié)果
2.1 基因突變數(shù)據(jù)收集和篩選
從dbSNP 數(shù)據(jù)庫中獲得TNNC1 基因SNP ID共1 559 個,經(jīng)過人工分級篩選后得到102 個nsSNPs,篩選及預(yù)測流程如圖1所示。
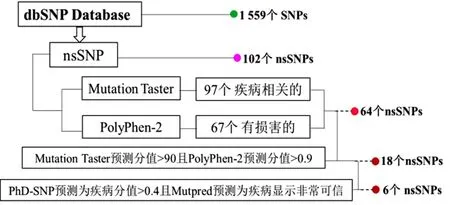
圖1 TNNC1 基因nsSNPs 篩選及預(yù)測流程Fig.1 Screening and prediction process of TNNC1 gene nsSNPs
2.2 突變位點有害性分析
利用Mutation Taster 及PolyPhen-2 有害性分析軟件,對在dbSNP 平臺上所篩選到的102 個nsSNPs進(jìn)行有害性分析。在Mutation Taster 軟件的預(yù)測結(jié)果中,3 個(I119V、I119T 和I119M)nsSNPs 被預(yù)測為多態(tài)性,2 個(I112M、E116D)nsSNPs 為未知意義,剩余97 個為疾病相關(guān)。而在PolyPhen-2 軟件的預(yù)測結(jié)果中,35 個nsSNPs 被預(yù)測為良性,其余67個均被預(yù)測為可能有害的。為了提高篩選的準(zhǔn)確性,發(fā)現(xiàn)被Mutation Taster 和PolyPhen-2 同時預(yù)測為有害的nsSNPs 為64 個。通過參考國內(nèi)外文獻(xiàn)發(fā)現(xiàn),在64 個有害的nsSNPs 中,A8V、A31S、E134D、C84Y 和D145E 在文獻(xiàn)中被報道為有害[5-6],與HCM 的發(fā)病是相關(guān)的,表明預(yù)測結(jié)果有較高的準(zhǔn)確性。研究發(fā)現(xiàn),蛋白保守序列對應(yīng)其重要的功能區(qū)域[12]。圖2為Mutation Taster 及PolyPhen-2 軟件對突變位點有害性及保守性分析,僅以rs897557713和rs1578263838 位點為代表進(jìn)行展示。從圖2(a)可以看出,rs897557713 位點預(yù)測為很可能有害的。從線蟲(celegans)、果蠅(dmelanogaster)、斑馬魚(drerio )、 非洲爪蟾 ( xtropicalis )、 小家鼠(mmusculus)、 原雞( ggallus)、 紅鰭東方鲀(trubripes)、貓(fcatus)、黑猩猩(ptroglodytes)和人類(human)等低等到高等物種間的氨基酸序列保守性分析發(fā)現(xiàn),該位點均為L(亮氨酸)較為保守,對維持該蛋白的功能十分重要,不可隨意改變,如圖2(c)所示。而rs1578263838 位點被預(yù)測為良性,從物種間的保守性分析發(fā)現(xiàn),該位點從低等到高等物種間的對應(yīng)氨基酸不保守可變,在不同物種間可為不同的氨基酸,如果蠅和紅鰭東方鲀的該基因位點上可為A(丙氨酸),線蟲上可為F(苯丙氨酸)。值得注意的是,在小家鼠和非洲爪蟾上可為M(甲硫氨酸),與突變體一致,因此該位點可變。當(dāng)由I(異亮氨酸)變?yōu)镸(甲硫氨酸)后,也能保持蛋白結(jié)構(gòu)和功能的完整性,不影響其生理生化功能。因此,結(jié)果表現(xiàn)為良性,如圖2(b)和(d)所示。
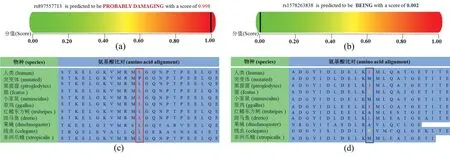
圖2 TNNC1 基因非同義突變位點的有害性(上)及物種間氨基酸保守性(下)分析。(a)rs897557713 位點;(b)rs1578263838 位點Fig.2 Damaging (the top)and amino acid conservation analysis of species (the bottom)were analyzed that non-synonymous mutations in TNNC1 gene. (a)rs897557713; (b)rs1578263838
2.3 突變位點疾病相關(guān)性分析
為了進(jìn)一步分析前期篩選的nsSNPs 與疾病的相關(guān)性,運用疾病關(guān)聯(lián)分析軟件PhD-SNP 和Mutpred,對前期Mutation Taster 預(yù)測分值>90(可信的)且PolyPhen-2 預(yù)測分值>0.9(很可能損傷的)的18 個(G159D、S69R、P52R、D149G、D3V、G140E、N51K、D151V、M47R、G110C、A23D、G140R、K158 N、C35Y、R147C、L48P、F74C 和V44G)nsSNPs 進(jìn)行下一步的分析。在分析結(jié)果中,18 個nsSNPs 均被PhD-SNP 軟件預(yù)測為疾病相關(guān)。同時,MutPred 軟件對18 個nsSNPs 預(yù)測的結(jié)果進(jìn)行顯示:這些突變位點改變了蛋白的理化特性、二級結(jié)構(gòu)或結(jié)構(gòu)域,其結(jié)果被認(rèn)為是可信或非常可信的,如表1所示。此外,對以上蛋白突變位點進(jìn)行結(jié)構(gòu)域定位發(fā)現(xiàn),除D3V、K158 N 和G159D 外,剩余15 個突變位點均位于蛋白質(zhì)PTZ00184 結(jié)構(gòu)域。在疾病相關(guān)性預(yù)測的結(jié)果中,其分?jǐn)?shù)與蛋白功能的重要程度呈正相關(guān)。在研究結(jié)果中發(fā)現(xiàn),PhD-SNP 軟件預(yù)測為疾病且可能性≥0.40 及Mutpred 軟件預(yù)測為非常可信的位點有6 個(G140E、D151V、G110C、K158N、G140R和L48P),這些位點的預(yù)測結(jié)果與疾病發(fā)生的關(guān)聯(lián)性和可信度是比較高的。如表1所示,G140E 突變導(dǎo)致氨基酸溶劑的可及性喪失,D151V 突變導(dǎo)致蛋白質(zhì)卷曲結(jié)構(gòu)功能喪失、G110C 突變改變了無序蛋白質(zhì)界面,K158N 突變改變蛋白卷曲結(jié)構(gòu),G140R突變改變氨基酸結(jié)合位點,L48P 突變導(dǎo)致蛋白質(zhì)內(nèi)部結(jié)構(gòu)折疊紊亂。以上結(jié)果可以看出,不同的突變位點對蛋白質(zhì)的損傷是不一樣的,有涉及蛋白結(jié)構(gòu)改變,也有結(jié)合位點功能喪失,或改變氨基酸的溶劑可及性。
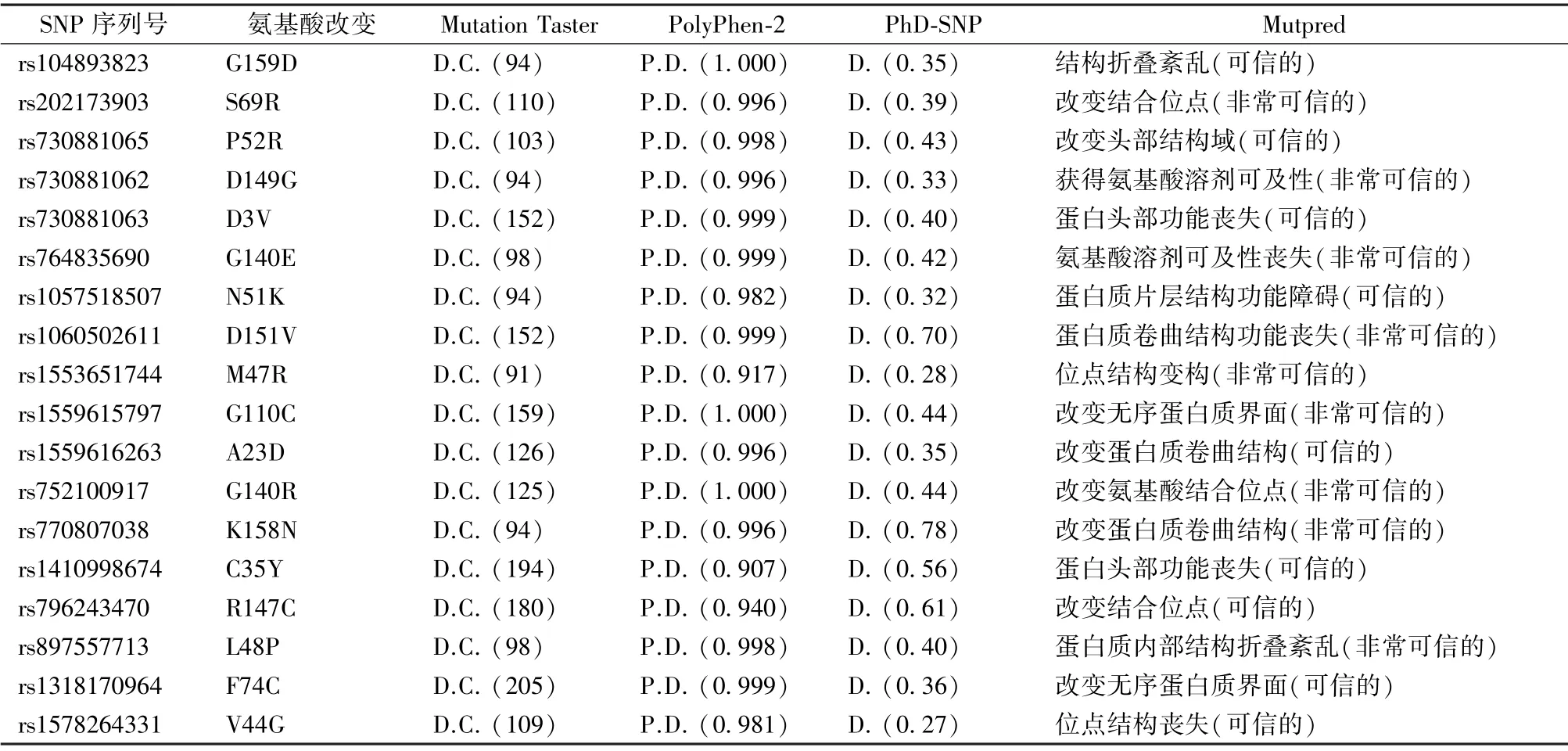
表1 nsSNPs 有害性及疾病關(guān)聯(lián)性分析Tab.1 Disease associations and harmfulness were analyzed of nsSNPs
2.4 致病相關(guān)突變位點結(jié)果分析及驗證
為了進(jìn)一步可視化分析與疾病相關(guān)的氨基酸突變位點,基于Swiss Model 數(shù)據(jù)庫進(jìn)行蛋白三維模型構(gòu)建。在模型構(gòu)建過程中,選擇序列相似性>99%和重合度>80%且評分最高的模板(RCSB PDB NO. 1AJ4),保證了建模的可信度。模型構(gòu)建完成后,利用VMD 軟件進(jìn)行可視化分析。在此僅展示代表性nsSNPs,圖3(a)表示突變前G(甘氨酸)為非極性脂肪族氨基酸,具有很強的親水性,溶于極性溶劑,多埋藏于蛋白質(zhì)內(nèi)部。圖3(b)表示突變后為E(谷氨酸),E 為不帶電荷的極性兼酸性氨基酸。氨基酸的極性與非極性、疏水性與親水性等性質(zhì),能影響氨基酸側(cè)鏈R 基團(tuán)的功能。
因此,G140E 突變位點對蛋白二級結(jié)構(gòu)的影響無明顯變化,但能影響蛋白質(zhì)氨基酸的溶劑可及性;圖3(c)和(d)為L48P 突變,L(亮氨酸)和P(脯氨酸)均為非極性氨基酸,但P 多藏于蛋白質(zhì)內(nèi)部。從圖中可以看出,突變前后二級結(jié)構(gòu)發(fā)生明顯變化,導(dǎo)致了蛋白質(zhì)內(nèi)部折疊發(fā)生紊亂,三維建模與Mutpred 軟件預(yù)測結(jié)果一致。
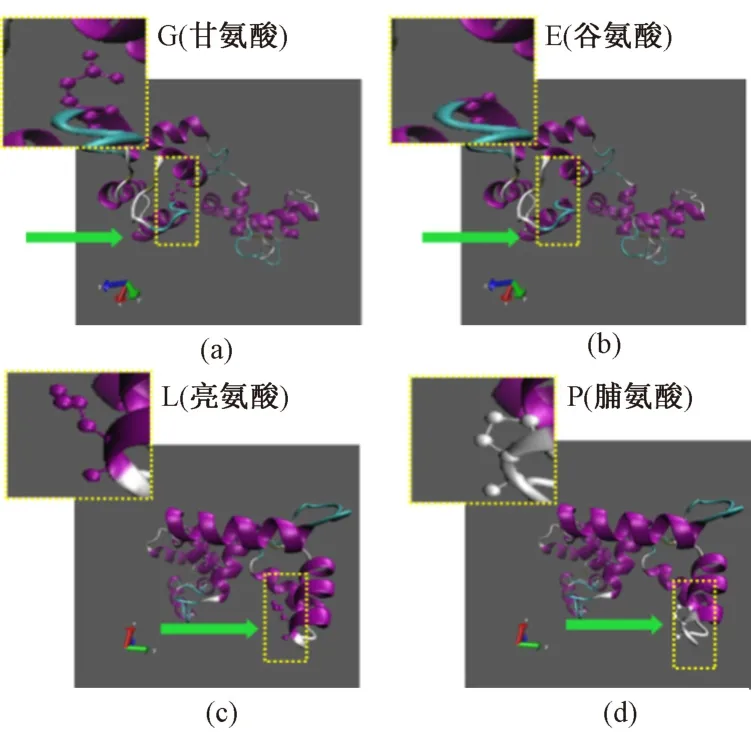
圖3 TNNC1 基因疾病關(guān)聯(lián)nsSNPs 位點野生型和突變型結(jié)構(gòu)模型預(yù)測(突變位點用黃色框標(biāo)注,并進(jìn)行放大)。(a)和(b)分別代表第140 位野生型G (甘氨酸)和突變型E (谷氨酸);(c)和(d)分別代表第48 位野生型L (亮氨酸)和突變型P(脯氨酸)Fig.3 The structure modeling of predicted pathogenic nsSNPs with wild-type and mutated in TNNC1 gene(Mutated site are emphasized by a yellow box and are locally zoomed). (a)and (b)indicates the position of 140 wild-type G ( glycine )and mutation E(glutamicacid),respectively; (c)and (d)indicates the position of 48 wild-type L (leucine)and mutation P (proline),respectively
3 討論
人類遺傳學(xué)領(lǐng)域的巨大挑戰(zhàn)之一,是明確SNPs與疾病之間的關(guān)系。在基因組中發(fā)現(xiàn)了超過1 000萬個SNPs,并且發(fā)現(xiàn)數(shù)量還在持續(xù)增加。因此,區(qū)分這些SNPs 對蛋白質(zhì)功能的影響,篩選出與疾病發(fā)生有關(guān)的遺傳變異位點,是實施精準(zhǔn)醫(yī)學(xué)和分子診斷的關(guān)鍵[13]。但結(jié)合臨床樣本,在實驗室對大規(guī)模的SNP 位點進(jìn)行功能分析,工作量巨大,且價格昂貴。因此,可以使用計算機分析工具,結(jié)合生物信息學(xué)手段,對可能有害的nsSNPs 進(jìn)行優(yōu)先級排序,找出可能與遺傳疾病發(fā)生相關(guān)的nsSNPs 位點,縮小研究范圍,降低研究成本,這是一種理想的研究策略[8]。
肥厚型心肌病是最常見的遺傳性心臟病,大部分發(fā)患者群常與肌節(jié)蛋白變異有關(guān)。TNNC1 作為編碼與Ca2+誘捕敏感蛋白的最重要基因之一,當(dāng)相關(guān)位點發(fā)生突變時,導(dǎo)致Ca2+循環(huán)紊亂,產(chǎn)生代償性的心肌細(xì)胞肥大,從而引發(fā)HCM[2]。生物信息學(xué)工具本身基于不同的數(shù)據(jù)庫和算法,靈敏度和準(zhǔn)確性有差異。在本研究的初級篩選過程中,使用到Mutation Taster 和PolyPhen-2 軟件聯(lián)合分級篩選,保證了致病相關(guān)位點預(yù)測的準(zhǔn)確性。Schwarz 等[14]的研究顯示,MutationTaster 軟件集成了來自不同生物醫(yī)學(xué)數(shù)據(jù)庫的信息,并使用了已建立的分析工具,分析了包括物種進(jìn)化保守性和蛋白質(zhì)特征喪失等,能較好地預(yù)測出潛在的有害變異位點。PolyPhen-2軟件集成了UCSC Genome Browser 的人類基因組注釋數(shù)據(jù)庫,能預(yù)測氨基酸改變后對人類蛋白質(zhì)結(jié)構(gòu)穩(wěn)定性和功能的可能存在影響[9]。在初級篩選的過程中發(fā)現(xiàn)的64 個有害性nsSNPs 中,包括了5 個已報道的nsSNP 位點(A8V、A31S、E134D、C84Y 和D145E)。Parvatiyar 等[6]在HCM 患者中發(fā)現(xiàn)了TNNC1 基因的A31S 突變,該突變增加了Ca2+的敏感性,促使心律失常,導(dǎo)致HCM 的發(fā)生。此外,Venter 等[15]用MutPred 軟件,成功預(yù)測了線粒體DNA 與復(fù)雜心血管疾病之間的關(guān)系。為了進(jìn)一步提高預(yù)測可靠性,在預(yù)測分析突變位點與疾病的相關(guān)性時,還聯(lián)合運用了PhD-SNP 和Mutpred 預(yù)測分析軟件,首次篩選出18 個致病相關(guān)的nsSNPs,這些位點從蛋白的理化特性、二級結(jié)構(gòu)或結(jié)構(gòu)域等方面影響了蛋白質(zhì)的功能。需要指出的是,各nsSNPs 具體的生物學(xué)功能及可能的疾病機制,還有待進(jìn)一步通過實驗深入研究。
4 結(jié)論
本研究基于多種生物醫(yī)學(xué)數(shù)據(jù)庫及平臺,以TNNC1 基因為示范,利用生物信息學(xué)方法,成功挖掘出TNNC1 基因中18 個與疾病相關(guān)的高風(fēng)險nsSNPs,為進(jìn)一步研究TNNC1 基因突變與HCM 的疾病表型關(guān)系打下理論研究基礎(chǔ)。同時,該方法也對其他遺傳疾病致病基因nsSNPs 與疾病關(guān)聯(lián)突變篩查具有重要的參考價值。
猜你喜歡
民用飛機設(shè)計與研究(2020年4期)2021-01-21 09:15:02
電子制作(2018年18期)2018-11-14 01:48:24
財經(jīng)(2017年15期)2017-07-03 22:40:49
財經(jīng)(2017年2期)2017-03-10 14:35:35
山東工業(yè)技術(shù)(2016年15期)2016-12-01 05:31:22
財經(jīng)(2016年15期)2016-06-03 07:38:02
財經(jīng)(2016年3期)2016-03-07 07:44:46
財經(jīng)(2016年6期)2016-02-24 07:41:51
中國中醫(yī)藥現(xiàn)代遠(yuǎn)程教育(2014年11期)2014-08-08 13:23:44
終身教育研究(2014年5期)2014-02-28 01:23:06
