單磷酸腺苷激活的蛋白激酶(AMPK):能量、葡萄糖感受器和代謝性疾病治療靶標
2022-05-20 07:08:56張宸崧王子涵陳燕雯萬芷辰王心茗陳思薇崔立楓
廈門大學學報(自然科學版) 2022年3期
關鍵詞:水平
張宸崧,王子涵,陳燕雯,萬芷辰,王心茗,陳思薇,崔立楓
(廈門大學生命科學學院,細胞應激生物學國家重點實驗室,福建 廈門 361102)
單磷酸腺苷激活的蛋白激酶(AMPK)是一種重要的調節細胞物質和能量代謝的蛋白質.早在1973年,人們便發現從大鼠肝臟中提取出來的一個具有激酶活性的組分,同時具有磷酸化并抑制乙酰輔酶A羧化酶(ACC)和3-羥基-3-甲基戊二酸單酰輔酶A還原酶(HMGCR)這兩個脂質合成關鍵蛋白的活性[1-2],后續研究表明該組分能夠被單磷酸腺苷(AMP)所活化[3-4].鑒于此,在1988年它被重新命名為AMPK[5].AMPK能通過磷酸化其下游的多種蛋白達到促進分解代謝、抑制合成代謝的效果,從而在生物體內發揮在能量水平上“開源節流”的作用.本文從AMPK的兩類調控方式入手,詳述其在代謝性疾病上“糾偏”的作用,并解釋其生理功能和作為藥物靶標的潛力.
1 AMPK的結構
如圖1所示,AMPK是一個異源三聚體,由α、β和γ 3種亞基組成.在哺乳動物中,α和β亞基分別有2種亞型[6-7],γ亞基有3種亞型[8],這就意味著理論上有至少12種AMPK蛋白結構.最新的研究表明,AMPK的3種亞基的不同亞型在表達和調控方式上具有時空特異性,這提示在不同的組織器官中,AMPK在調節方式和下游效應上可能存在著差別[9-10],也提示以AMPK為靶點的藥物開發可能需要注意這種特異性[11].
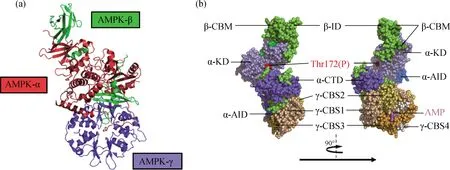
圖(a)數據來自PDB 4cff;圖(b)數據來自PDB 6c9j;KD.激酶結構域;AID.自抑制結構域;CTD.羧基端(C端)結構域;CBM.碳水化合物結合模塊;ID.交互結構域;CBS.胱硫醚β合成酶序列;Thr172(P).蘇氨酸172(磷酸化).
AMPK的α亞基是重要的激酶活性亞基,主要由α-KD、α-AID和α-CTD組成.α-AID的存在會抑制α-KD 的激酶活性[12];在α-KD的C端小葉上有一個重要的位點——Thr172,AMPK的上游激酶對該位點的磷酸化能夠解除α-AID的自抑制作用,是最大程度地激發AMPK活性所必需的[12-13].
AMPK的β亞基結構尚未完全解析,主要包括氨基端(N端)的豆蔻酰化區域、β-CBM和β-ID.β亞基N端的豆蔻酰化修飾對AMP和二磷酸腺苷(ADP)促進AMPK的Thr172位點磷酸化有關鍵作用[14-15],而β-CBM本身則能和糖原等營養物質結合并調節AMPK[16].CBM還能和α亞基共同作用,在交界處形成變構藥物和代謝物(ADaM)位點,是重要的藥物靶向變構激活AMPK的位點[17].
AMPK的γ亞基含有4個CBS串聯重復序列,可以與AMP、ADP或三磷酸腺苷(ATP)結合,影響AMPK的構象.關于AMPK結構的更多細節和最新進展可參閱相關綜述文獻[18].
2 AMPK的調控機制
2.1 經典機制:通過AMP/ATP比值調控的變構激活
如上所述,AMPK發現的過程是和細胞內低能量水平的表征分子AMP緊密聯系在一起的,因此和AMP等腺苷類物質相關的激活被稱為經典機制.在細胞中,ATP是生物體內能量的“通貨”,ADP是ATP供能后失去一個磷酸基團的產物,AMP則是ATP失去一個焦磷酸基團或ADP失去一個磷酸基團的產物.在一般生理條件下,這3種物質的比例處于一種比較穩定的狀態;而當機體處于過度饑餓、缺血或者其他極端條件下時,ATP產生不足而ADP累積,此時機體通過2ADP → ATP + AMP反應在一定程度上彌補ATP的不足,因而導致AMP的水平升高[19].AMPK正是通過其γ亞基上的腺苷酸結合位點來感應這3種腺苷酸的水平:該位點由4個高度相似的CBS區段——CBS1、CBS2、CBS3和CBS4組成,除CBS2外,另3個區段都可以結合腺苷酸,且CBS1和CBS4對腺苷酸的結合還能加強CBS3結合AMP的能力[20],使AMPK感應AMP的能力較ADP和ATP靈敏得多[21].當機體AMP含量較高,即能量水平較低時,CBS3與AMP結合,使γ亞基發生變構,這種變構調節導致α亞基的Thr172位點暴露,進而促進AMPK的上游激酶對其的磷酸化,解除AID對KD的抑制作用,從而提高AMPK的活性[22-23].除AMP外,ADP也能結合CBS3,但其作用尚存在爭議[15,21,24];而當機體內ATP含量較高即能量水平較高時,CBS3則被ATP占據,使AMPK保持在利于Thr172位點被去磷酸化的構象,引起其活性的抑制[20,25].AMPK正是借此感應細胞內的能量水平,并在低能量水平下被激活,進而調節一系列下游信號通路,抑制合成代謝、促進分解代謝,以維持正常的機體能量水平.生化實驗證據顯示AMP/ATP比值隨ADP/ATP比值的平方而變化[19],故而感知AMP相較于ADP更靈敏.加之不同γ亞基的亞型感應AMP的能力不同,如在體外γ1和γ2感應AMP的能力顯著強于γ3[26],且如前所述不同亞型的組織和細胞的時空特異性分布不同,這些機制很可能保證了細胞內的能量變化被靈敏地感知并被嚴謹地調控.
體外實驗結果表明,Thr172位點的磷酸化可使AMPK活性提升100倍以上[27],因此被認為是AMPK最重要的活性調節方式.目前已發現的可以磷酸化Thr172位點的上游激酶主要有3種,其中和感應能量水平即AMP、ADP調控相關的是肝臟激酶B1(LKB1).它于1998年在波伊茨-耶格綜合征中被發現[28],并隨后被證明對Thr172位點的磷酸化起主要作用[29-31].盡管LKB1和AMPK是激酶與底物的關系,但AMP或ADP促進的LKB1對AMPK的磷酸化卻不僅需要這兩個分子本身,還需要更多胞內結構的參與:原核表達的AMPK在體外(游離狀態)無法重現AMP、ADP的這一作用[32].而在1997年,Mitchelhill等[33]發現AMPK的β亞基N端能被豆蔻酰化修飾,提示AMPK在胞內是能夠結合到細胞膜系統上的;2010年,Oakhill等[14]進一步發現,β亞基N端第2位甘氨酸位點的豆蔻酰化修飾能夠促進AMPK結合到膜上,且這是AMP、ADP激活AMPK所必需的:通過對原核表達的AMPK進行豆蔻酰化修飾,就能使其響應AMP、ADP,促進LKB1對Thr172位點的磷酸化;此外,使用依賴于糖酵解產能(葡萄糖饑餓后能引起AMP升高)的COS7細胞系,在葡萄糖饑餓的條件下,在細胞水平驗證了AMPK膜定位的重要性.
AMP對AMPK的變構激活還體現在其能使被磷酸化后的AMPK活性進一步提升2~3倍(隨ATP水平而變[21]),達到最大程度的激活[34].AMP和ADP的結合還有抑制AMPK被磷酸酶去磷酸化的作用[24,35],最新研究表明這其中還需要多個分子調節AMPK出入細胞核[36].
AMPK的變構調節機制是設計靶向AMPK的小分子藥物的重要依據.以MK-8722(靶向ADaM位點)[37]等藥物為代表的AMPK變構調節激活劑[38],被證明可以有效避免基于抑制線粒體ATP產生以提高AMP水平的機制所設計的藥物導致的細胞毒性[39-40],因而具有很好的應用前景;但這類化合物目前需要解決的關鍵問題是如何避免無差別、強烈地全局性激活體內AMPK所引起的副作用.
2.2 非經典機制:通過鈣離子或對葡萄糖水平感應的激活
除依賴AMP、ADP之外的AMPK激活方式被統稱為非經典機制.其中,鈣調素依賴蛋白激酶的激酶β(CaMKKβ)能被胞內大量鈣離子水平升高所激活(通過結合鈣調蛋白,后者在鈣離子結合的情況下結合CaMKKβ,使其自抑制作用被解除),再引起AMPK的Thr172位點磷酸化,因而在神經等系統中發揮重要作用[41].DNA損傷所引起的AMPK激活也被認為由CaMKKβ介導[42].而轉化生長因子β活化激酶1(TAK1)則是一種有絲分裂原活化蛋白激酶的激酶(MAPKK),在內吞體/溶酶體損傷、有絲分裂原刺激等條件下能夠引起AMPK的激活[43-44].現有證據表明,至少在體外的單一體系下,CaMKKβ和TAK1對AMPK的激活獨立于AMP依賴的AMPK激活途徑[44-45],盡管在特定的生理條件下兩者之間可以交互影響[43].除CaMKKβ和TAK1介導的非經典激活外,靶向ADaM位點的藥物如MK-8722、水楊酸和A769662,則能夠通過抑制Thr172位點的去磷酸化以及變構調節的方式激活AMPK[46-48].
AMPK的另一類非經典激活方式則與機體感應葡萄糖水平的降低有關,它是AMPK激活最普遍的情況,不僅常見于如餐間饑餓、運動等多種生理條件下,而且從酵母到哺乳動物高度保守[49].盡管葡萄糖本身作為機體主要的能量來源和ATP的產生有密切聯系,但與COS7細胞系以及許多腫瘤細胞系不同,本課題組及相關研究都發現,在肌肉、肝臟等不依賴于糖酵解為主要ATP生產方式的正常組織中,葡萄糖水平的下降并不能引起AMP水平的上升[50-51],這很可能是由于機體和組織內豐富的氨基酸(如谷氨酰胺)、脂肪酸等替代碳源能夠在葡萄糖水平下降時及時補充并生產ATP[52].本課題組發現:此時AMPK的激活需要在溶酶體膜表面進行,依賴于體軸發育抑制因子(AXIN)、定位在溶酶體膜上的Ragulator復合體和v型ATP酶(v-ATPase)的參與.當葡萄糖水平下降時,v-ATPase被抑制,它和Ragulator復合體發生構象變化,促進AXIN與之結合并被招募到溶酶體附近.AXIN則可以結合LKB1,作為“橋梁”連接定位在溶酶體附近的AMPK和LKB1,從而促進LKB1對Thr172位點的磷酸化,最終在溶酶體附近激活AMPK[53-54].早在這一機制發現之前,已有研究通過蛋白質組學手段鑒定AMPK能定位在溶酶體上[55],且本課題組發現的AMPK激活機制也依賴于β亞基的豆蔻酰化,因此將上述機制稱為“溶酶體途徑”[54](圖2).需要注意的是,相較于豆蔻酰化,盡管β亞基的CBM與糖原對AMPK的調節有關,但卻并不參與葡萄糖對AMPK的調節[56],ADaM位點也不參與該過程[51].有意思的是,Ragulator早先被認為是細胞內另一個重要代謝調節者——雷帕霉素靶蛋白復合體1(TORC1,在哺乳動物中稱為mTORC1)維持活力所必需的[57-58];與AMPK相反,mTORC1在細胞內能量和物質充足時遷移到溶酶體表面被激活并啟動合成代謝,促進細胞增長和增殖[59].可見AMPK和mTORC1的激活對應于不同的營養、能量條件,卻“借道”同一類分子行使調控作用,這可能是一種簡約而又精確的調節方式.
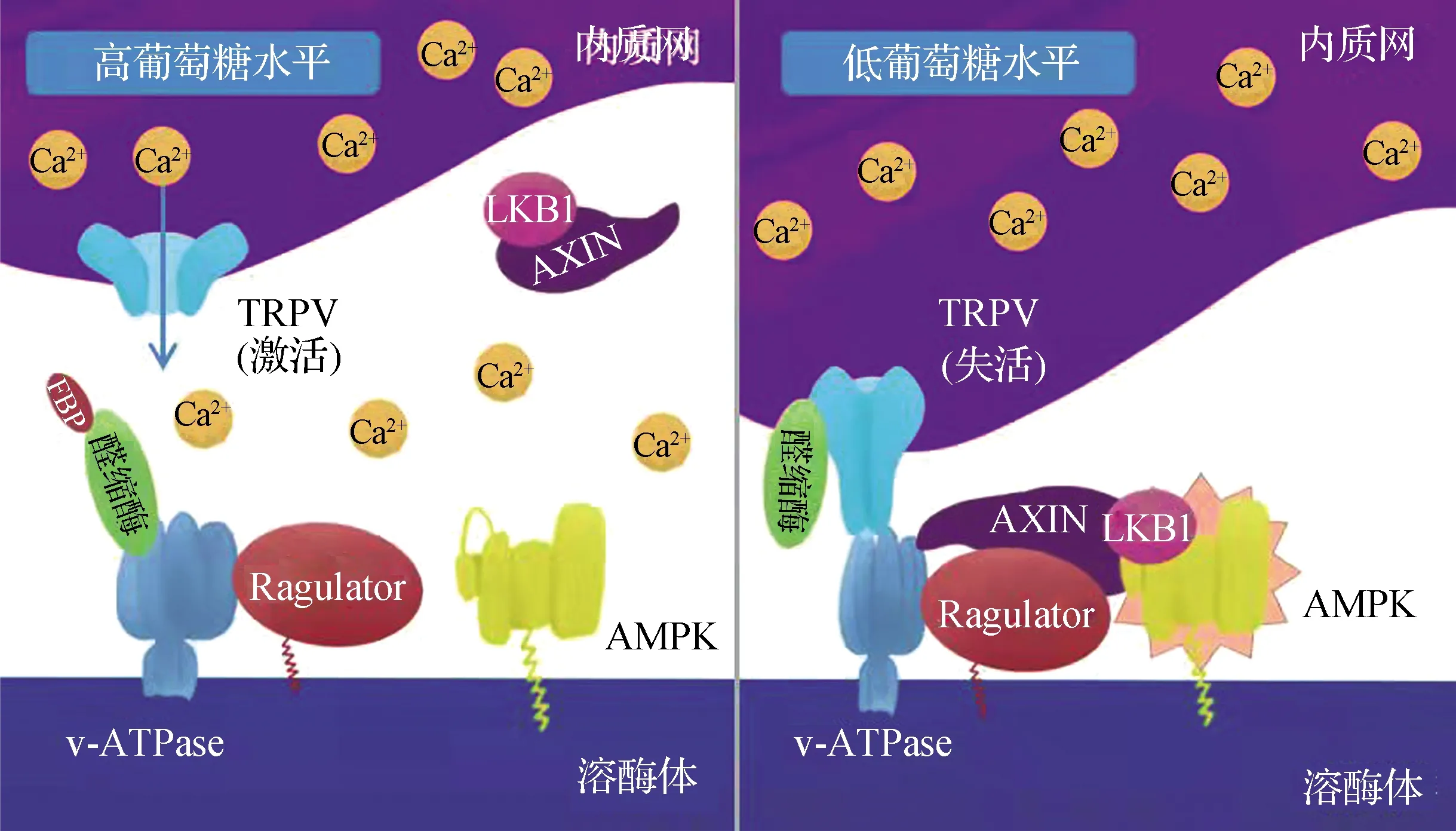
FBP.果糖-1,6-二磷酸.
葡萄糖水平下降能夠在不依賴AMP的基礎上激活AMPK,這說明它有另一套信號被感知以調控AMPK.本課題組發現糖酵解途徑的中間產物FBP是介導這一過程的關鍵信號分子[51].FBP在葡萄糖水平下降時下降,不再結合催化它形成三碳糖的醛縮酶,進一步導致v-ATPase-Ragulator復合體的變構[60](圖2),可見醛縮酶是感受FBP乃至葡萄糖水平的感受器.作為糖酵解途徑最直接(沒有旁路)的產物之一,FBP的產率常被認為是糖酵解速率的表征[61],因此它比AMP更靈敏,在細胞AMP水平升高前AMPK即可通過感知機體葡萄糖水平的降低而被激活,“前瞻性”地調節細胞代謝穩態.有意思的是,本課題組在體外單純含有v-ATPase和醛縮酶的體系中,無法觀察到FBP對v-ATPase的調節,這強烈暗示還有其他因子參與該過程;進一步鑒定發現陽離子通道瞬時受體電位香草酸亞型(TRPV)是介導這一過程的關鍵.TRPV主要定位于細胞膜和內質網上,其中有一部分內質網定位的TRPV能夠在內質網-溶酶體接觸的位置和溶酶體途徑產生聯系:低葡萄糖水平下,未結合FBP的醛縮酶結合并抑制TRPV的活性,抑制其從內質網釋放鈣離子的作用,從而降低內質網-溶酶體接觸位置的鈣離子濃度,使得TRPV結合v-ATPase從而抑制其活性[60](圖2).需要注意的是,這里所提到的鈣離子,并非是前述能激活CaMKKβ所釋放的大量鈣,而是原本很低濃度的鈣離子濃度進一步降低所引起的情況,對AMPK的調節作用也并不同于前者.
在分子機制上,溶酶體途徑和經典機制是相互獨立的,即AMP可以繞過溶酶體途徑對AMPK行使激活的功能,而溶酶體途徑可以不需要AMPK結合AMP,兩者并不互相影響;在層級關系上,經典機制又是溶酶體途徑的上級,能造成比溶酶體途徑更強、更廣泛的AMPK激活.這進一步體現在不同激活方式下AMPK所具有的時空特異性調控特點上:當葡萄糖濃度下降(AMP水平還未上升)時,溶酶體上的AMPK先經溶酶體途徑被激活;當AMP水平上升時,如前述更加嚴重的營養缺失發生時,胞質中的AMPK也開始逐漸被激活,此時AXIN通過直接加強LKB1和AMPK的相互作用介導該過程[53];而當細胞中能量嚴重匱乏,如在缺氧、缺血等情況發生時,線粒體上的AMPK最終被激活,這只需要AMP的參與.鑒于能量嚴重匱乏的情況并非生理常態,該機制可能部分解釋了為何直接靶向AMPK,以及以引起全局性AMPK激活為策略所設計的藥物往往難以避免一些副作用,如MK-8722所引起的心肌肥大[37],后者最近被證實確實是由于心臟中AMPK過度激活所導致的[62].因此,基于葡萄糖感應的溶酶體途徑也許可為針對不同代謝疾病設計相應的引起AMPK激活的藥物提供新思路.
3 AMPK的底物及其對代謝的調控
如上所述,AMPK在功能上可以概括為“促進分解代謝,抑制合成代謝”,從而在營養和能量匱乏的情況下維持機體的物質與能量穩態.AMPK通過磷酸化一系列底物直接或間接行使這些功能[63](圖3).
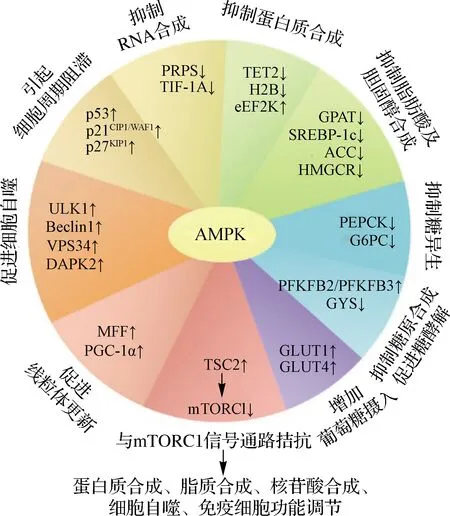
TET2.Tet甲基胞嘧啶雙加氧酶2;H2B.組蛋白2B;eEF2K.真核延伸因子2激酶;GPAT.甘油-3-磷酸酰基轉移酶;SREBP-1c.固醇調節元件結合蛋白-1c;PEPCK.磷酸烯醇式丙酮酸羧激酶;G6PC.葡萄糖-6-磷酸酶催化亞基;PFKFB.6-磷酸果糖-2-激酶/果糖-2,6-二磷酸酶;GYS.糖原合酶;GLUT.葡萄糖轉運蛋白;TSC2.結節性硬化癥復合體亞基2;MFF.線粒體裂殖因子;PGC-1α.過氧化物酶體增殖物激活受體-γ共激活因子-1α;ULK1.Unc-51樣自噬啟動激酶1;Beclin1.自噬效應蛋白1;VPS34.分揀蛋白34;DAPK2.死亡相關蛋白激酶2;PRPS.磷酸核糖焦磷酸激酶;TIF-1A.轉錄中介因子-1α;p53、p21CIP1/WAF1、p27KIP1均為細胞命運決定相關蛋白.
3.1 AMPK調節營養物質代謝
AMPK對糖、脂質、核苷酸和蛋白質等已知的所有種類的營養物質都具有調節作用.在糖代謝方面,AMPK促進葡萄糖的分解,抑制葡萄糖的生成和儲存.AMPK可以磷酸化并抑制GYS,抑制糖原合成[64-65];也可磷酸化磷酸果糖激酶PFKFB2和PFKFB3,促進糖酵解[66-67];還可以促進葡萄糖轉運至細胞內以維持其分解,如分別通過磷酸化Tre-2/USP6-BUB2-cdc16結構域家族成員1(TBC1D1)和硫氧還蛋白互作蛋白(TXNIP),促進GLUT4和GLUT1轉移到細胞膜上[68-69],或通過轉錄調節因子肌細胞增強因子2(MEF2)和組蛋白脫乙酰基酶5(HDAC5)提升GLUT4的表達水平[70-71].在促進糖酵解的同時,AMPK也抑制糖異生作用,如通過磷酸化初乳堿性蛋白(CBP)、環磷腺苷效應元件結合蛋白調控的轉錄共激活因子2(CRTC2)、HDACs(及其下游的叉頭框蛋白O(FOXO))、碳水化合物反應元件結合蛋白(ChREBP)等一系列轉錄因子和轉錄調節因子調節其轉錄活性,或直接調節干細胞核因子4α(HNF4α)等轉錄因子的表達水平,抑制PEPCK、G6PC等糖異生相關限速酶的表達[72-76].需要注意的是,AMPK相關激酶(ARK)家族的其他蛋白也能行使AMPK抑制糖異生的功能(如對CRTC2和HDACs的磷酸化)[77],從而保證糖異生這一高度耗能的過程在營養和能量水平較低時被準確地調控.
脂質合成是高度耗能的代謝過程,而它本身作為機體重要的儲能物質,則能夠被分解而產生大量ATP.作為其經典功能之一,AMPK通過多個底物促進脂質的攝取和分解,抑制脂質合成.AMPK可以磷酸化并抑制ACC(包括定位于細胞質的ACC1[1]和線粒體的ACC2[78])的活性,減少丙二酸單酰輔酶A的產生,而后者的產生既是脂質從頭合成(DNL)途徑的限速步驟,本身又能夠抑制線粒體進行脂肪酸的攝取和β-氧化[1,79].同樣地,AMPK對HMGCR的磷酸化和抑制也是抑制膽固醇合成的關鍵[2].與糖代謝類似,AMPK還能促進脂肪酸轉運體CD36向細胞質膜的轉移,促進細胞攝入脂肪酸[80].AMPK還可以磷酸化轉錄因子SREBP-1c并抑制其被蛋白酶降解,后者是調控包括ACC、脂肪酸合成酶(FAS)等DNL關鍵基因轉錄的因子[81].類似地,AMPK也能通過上述磷酸化ChREBP的機制,從轉錄水平抑制DNL[82].除脂肪酸合成和β-氧化外,AMPK還能通過磷酸化GPAT,抑制甘油三酯(TAG)和磷脂合成的共同底物——甘油二酯(DAG)的生成,從而抑制TAG和磷脂的合成[83].
盡管被認識得較晚,但已有多項研究表明,與糖類、脂質類似,核酸也是重要的儲能物質,且其合成也是高度耗能的.如在胸腺細胞中,RNA的合成至少消耗了15%的有氧呼吸產能[84],而作為細胞內核酸的儲存庫之一,核糖體(儲存體內80%以上的核糖體RNA)能夠在營養物質缺乏時被降解以產能[85].現有研究表明AMPK能夠抑制核酸的合成,如:AMPK能磷酸化并抑制嘌呤合成途徑的限速酶PRPS,從而抑制嘌呤核苷酸的從頭合成[86];AMPK也能磷酸化并抑制調控RNA聚合酶Ⅰ轉錄的關鍵轉錄因子TIF-1A,從而抑制RNA的合成[87].
蛋白質的合成也是機體內主要的耗能過程,類似于核酸,胸腺細胞中的蛋白合成消耗了約20%的有氧呼吸產能[84],AMPK則能通過多種方式抑制蛋白質的合成,促進蛋白質的分解.AMPK最主要的作用是通過抑制mTORC1阻止蛋白質的翻譯起始:AMPK能夠磷酸化TSC2并激活TSC1/TSC2/TBC1D7復合體,后者作為mTORC1的激活因子——小G蛋白Rheb的三磷酸鳥苷酶活化蛋白,能夠直接抑制mTORC1活性[88-89];AMPK也可以磷酸化mTORC1復合體上的Raptor亞基從而直接抑制mTORC1活性[90].需要注意的是,除促進蛋白質的翻譯外,mTORC1也能促進脂質(如SREBP1[91]和HMGCR[92])和核苷酸(包括嘌呤[93]和嘧啶[94-95])的合成,因此AMPK通過mTORC1調節機體代謝的方式可謂“牽一發而動全身”.另外還需要說明的是,和前文溶酶體途徑中提到的AMPK與mTORC1響應葡萄糖水平并經共同因子在溶酶體上被調控的作用不同,AMPK對于mTORC1的抑制僅限于其活性而不涉及mTORC1的溶酶體定位,它起到的是快速而靈敏地抑制mTORC1的作用:當AMPK缺失時,mTORC1仍能響應葡萄糖水平下降,離開溶酶體表面從而被抑制,但速度明顯變慢[96].除mTORC1外,AMPK還可以通過磷酸化并激活eEF2K從而抑制多肽鏈的延伸[97],或通過磷酸化并抑制組蛋白H2B和雙加氧酶TET2在轉錄水平上抑制蛋白的表達[98-99].
3.2 AMPK調節細胞代謝
除調節糖、脂質等具體代謝途徑外,AMPK還能在細胞水平上整體性地調節代謝過程,包括促進自噬作用、維持線粒體的質量和數量以及調節細胞周期等過程.
自噬作用,特別是巨自噬作用,是細胞無差別降解生物大分子乃至包括線粒體、脂滴、核糖體、內質網等細胞結構,維持能量產生和營養物質釋放與再利用的關鍵過程.AMPK的激活可通過以下至少4個方面的機制顯著促進細胞的自噬作用:1)AMPK可以磷酸化啟動自噬的關鍵蛋白ULK1的多個位點,激活ULK1,從而啟動自噬[100-101];2)ULK1也受到mTORC1的結合與抑制,而AMPK對mTORC1的抑制能夠解除mTORC1對ULK1的抑制作用,從而激活ULK1[101];3)AMPK可以磷酸化自噬調節相關蛋白Beclin1和VPS34,也可與ULK1協同調節VPS34,從而促進VPS34和Beclin1共同組成自噬啟動復合體,加快自噬[101-102];4)AMPK還能夠磷酸化并激活DAPK2,促進DAPK2對Beclin1的磷酸化從而促進自噬作用[103].
AMPK對線粒體的作用可歸納為“除舊迎新”,即通過清除細胞內已損壞、呼吸效率低下(如由呼吸作用所產生的活性氧(ROS)而造成膜損壞,引起質子泄漏和質子梯度部分喪失等)的線粒體,促進新的線粒體生成.在清除受損線粒體方面,AMPK可以通過加強自噬作用促進受損線粒體的降解[100];同時,AMPK能夠通過磷酸化MFF促進線粒體的分裂,使得受損的線粒體更易分裂從而經自噬作用被降解[104].而在促進線粒體生成方面,AMPK能夠磷酸化并激活PGC-1α的轉錄活性,后者是介導線粒體生成所需核編碼基因的轉錄所必需的[105].AMPK也能夠通過上調輔酶Ⅰ(NAD+)的水平,促進NAD+調節的去乙酰化酶Sirtuins對PGC-1α的去乙酰化,進一步上調PGC-1α的轉錄活性[106].此外,上述AMPK對合成代謝的抑制作用(如ACC調節的脂肪酸合成)在維持能量平衡的同時減少細胞內的還原力消耗,從而維持胞內ROS水平即氧化還原穩態,以保護線粒體結構和功能的完整性[107];同時,AMPK也能夠通過FOXO、核因子E2相關因子2(NRF2)等轉錄因子,在轉錄水平上維持細胞中抗氧化蛋白的表達水平[108-109].特別地,AMPK還能夠通過上述加強糖、脂質等分解代謝途徑的方式,瞬時性地提升線粒體的有氧呼吸強度(該作用被稱為“mitohormesis”),也能進一步激活PGC-1α,促進線粒體生成[110].AMPK的上述作用使得胞內線粒體具有更高的呼吸效率,在產生等量ATP的同時,減少質子的泄漏和ROS的產生.總體上看,AMPK的這一作用可促進細胞代謝模式轉向有氧呼吸,這也是后文所述其抑制炎癥、癌癥、延長壽命等功能的重要依據[111].
此外,AMPK能引起細胞周期的阻滯,暫停細胞分裂過程中眾多合成代謝過程所引起的能量消耗,例如:AMPK能夠磷酸化并激活p53,通過其下游周期細胞依賴性激酶(CDK)的抑制因子p21CIP1/WAF1阻滯細胞周期于G1期[112];AMPK也能夠磷酸化另一CDK的抑制因子p27KIP1,介導G1期阻滯[113];除G1期阻滯外,在成熟紅細胞中還發現AMPK能夠阻滯細胞周期于S期,并可誘導細胞凋亡,從而可能在維持紅細胞更新的過程中發揮重要作用[114].AMPK在調節細胞周期方面的作用也是后文所述的抑制癌癥相關功能的依據.
3.3 AMPK與免疫反應
近年來隨著代謝與免疫的聯系被不斷揭示,傳統上的免疫反應被更廣泛地認為是一種代謝所介導的過程[115];而AMPK在這一過程中,特別是在調節炎癥相關免疫反應中起到了關鍵作用,該作用可以看作AMPK調節一類特化細胞的代謝所介導的功能[116].目前,已發現AMPK能通過調節巨噬細胞、胸腺依賴性淋巴細胞(T細胞)、中性粒細胞和樹突狀細胞等免疫細胞類群,抑制炎癥反應的發生和發展.
AMPK能夠引起巨噬細胞從M1促炎類群轉變為M2抗炎類群[117].雖然具體分子機制尚不明確,但是已有研究發現,AMPK的這一功能可能和其改變了M1巨噬細胞的代謝模式而使之接近于M2巨噬細胞有關[116].在M1巨噬細胞被活化(即“經典激活”,如經歷脂多糖(LPS)或干擾素γ刺激)時,細胞的代謝模式轉向有氧糖酵解并加強戊糖磷酸途徑,而氧化磷酸化則被抑制且變得不完整[118-119],這一作用可能是M1巨噬細胞中促炎因子的生物合成所必需的[115](類似于后文所述的腫瘤細胞),且其產生的高濃度ROS和NO(由不完整的電子傳遞鏈介導[120-121])則進一步輔助其殺滅病原體[122-123].其中,M1巨噬細胞中PFKFB3的表達水平升高和Toll樣受體4(TLR4)所激活的mTORC1有關,后者進一步引發缺氧誘導因子1α(HIF1α)的表達水平上升.HIF1α原本被發現是在缺氧的條件下穩定表達,使乳酸脫氫酶 A和磷酸肌醇依賴性激酶1(PDK1)等基因表達上調,提升糖酵解產能以緩解缺氧所引起的有氧呼吸產能不足;而在這里它則通過同樣的方式促進有氧糖酵解[118,124].同時,NO的產生則破壞了電子傳遞鏈[119].如3.1小節所述,盡管AMPK能激活PFKFB3,但其對mTORC1的抑制起主導作用,因此整體來說AMPK抑制有氧糖酵解[125],而上述AMPK通過NAD+、Sirtuins和PGC-1α促進線粒體生成的作用也能夠維持氧化磷酸化.此外,Sirtuins還能夠去乙酰化并抑制核因子κB(NFκB)途徑中的p65,阻止促炎細胞因子的轉錄[126].而在M2巨噬細胞被活化(即“非經典激活”,如受到白介素-4(IL-4)刺激)的過程中,氧化磷酸化水平則急劇升高[127].AMPK不僅可以促進IL-4通過信號傳導及轉錄激活蛋白6(STAT6)介導的、由PGC-1α行使的線粒體生成,還能夠通過促進脂肪酸β-氧化給M2巨噬細胞提供能量,從而促進該細胞的活化[128-129].
AMPK也能調控T細胞的命運決定.類似于巨噬細胞,T細胞的分化也伴隨著代謝模式的改變.如CD8+T細胞被抗原呈遞細胞(APC)結合后開始分裂,APC附近的CD8+T細胞進行有氧糖酵解,分化成效應T細胞;遠端的則傾向于氧化磷酸化,分化為記憶T細胞[130-131].AMPK介導的mTORC1-HIF1α的抑制作用可抑制效應T細胞的分化而促進記憶T細胞的分化[132-133].類似地,CD4+T細胞在分化成輔助性T細胞的過程中,也需要mTORC1-HIF1α所介導的有氧糖酵解加強,從而行使類似于M1巨噬細胞的促炎作用;相反地CD4+T細胞分化成調節性T細胞則需要提升氧化磷酸化的水平[134-136],而AMPK的存在可促進調節性T細胞的形成[137].類似于后文所述的長壽相關代謝表型,T細胞分化中的這種代謝模式改變,可能與其在體內存活時間的長短以及對合成代謝的不同需求有重要聯系[138].
與M1巨噬細胞類似,中性粒細胞也能在LPS等刺激下由TLR4介導促炎癥過程,而AMPK的激活則可以逆轉該過程[139];同樣地,樹突狀細胞的活化和成熟也需要有氧糖酵解的加強,而AMPK則可以抑制這一過程[140].
綜上,AMPK通過對免疫細胞代謝模式的調節,影響了這些細胞的分化、成熟與功能.在大多數促炎癥反觸發的過程中,AMPK被顯著抑制(如LPS[141]),因此靶向AMPK的激活以抑制炎癥反應的策略,將有助于機體抵抗和有序地清除病原體的侵染(最新進展請參閱綜述文獻[142]),以及以慢性炎癥反應為特征之一的糖尿病、肥胖和癌癥的治療(見后文詳述).
需要特別說明的是,上述3個方面的AMPK對細胞的代謝調節作用是簡單基于AMPK在生理上的激活后進行的相關觀察,然而正如在2.2小節中所述,AMPK的激活在生理上隨著能量應激強度的升高,從葡萄糖缺乏到AMP上升,呈現出累進式、區域化的激活[143].且不同區域的AMPK激活能夠引起不同底物的磷酸化,并引起不同的代謝調節過程.如ACC2和MFF在葡萄糖缺乏的情況下不被磷酸化,而只能在AMP水平升高的情況下被線粒體上的AMPK磷酸化,這暗示著線粒體上AMPK底物所介導的代謝調節過程只能發生在嚴重的能量匱乏情況下[143].最近,該過程也被證明存在于動物體內[144].而前述DNA損傷所引起的CaMKKβ介導的AMPK激活則僅局限于細胞核內,AMPK也不能夠在此時磷酸化ACC等胞質中的蛋白,可見上述AMPK-p53所介導的細胞周期阻滯和AMPK對代謝途徑的調節也是相互平行的過程[42].因此,未來對AMPK在不同時空條件下的功能進行更深入的研究,將有助于針對不同的情況設計相應的藥物,以更好地干預和治療各種代謝性疾病.
4 AMPK和代謝性疾病——短期效應
上述AMPK在代謝調控中行使維持代謝穩態的功能,對由代謝紊亂特別是合成代謝過度活化的疾病(如肥胖所引起的Ⅱ型糖尿病、脂肪肝)乃至癌癥的癥狀緩解與預防有重要意義,也因此成為代謝性疾病治療的靶標.相較于后文所述對壽命等相對長期的作用,AMPK對代謝性疾病的作用可歸納為它的短期效應.
4.1 AMPK與肥胖相關代謝性疾病
由營養過剩引起的肥胖及其引發的Ⅱ型糖尿病、脂肪肝等并發癥,是當今最具代表性且對人類健康造成日趨嚴重威脅的代謝性疾病[145],而AMPK對代謝調節的功能在肥胖(包括內臟脂肪積累)到糖尿病(胰島素抵抗)發展過程中的多個紊亂代謝活動起到“糾偏”作用.目前的觀點認為,營養過剩引起的脂肪累積首先導致脂肪細胞TAG增多和肥大,引起細胞氧氣通透變差且消耗增加,造成相對的缺氧狀態,進而觸發HIF1α的上調,導致趨化因子分泌并吸引巨噬細胞、肥大細胞的聚集與活化,增加多種促炎細胞因子的分泌[146-147],后者又激活脂肪細胞中的NFκB和c-Jun N端激酶(JNK)信號通路,導致胰島素受體(IR)及其底物(IRS)的絲氨酸磷酸化,引發其失活和胰島素通路的抑制[148-149],該作用進一步通過血液擴展到肝臟、肌肉等外周組織,從而引起外周胰島素抵抗[150-152].除脂肪細胞肥大外,脂肪合成的增加也改變了內質網的脂成分,引起內質網應激,后者進一步提升局部炎癥反應[153-155].同時,脂肪細胞中TAG合成的增強導致中間產物DAG水平的升高,后者引起細胞內磷酸激酶Cε(PKCε)的激活,加強IR的蘇氨酸磷酸化而導致胰島素通路的抑制(實際上該作用也發生于肝臟(即脂肪肝)和肌肉中,且在肌肉中還有PKCθ參與行使類似的作用)[156-157];TAG合成的增強也導致血液中游離脂肪酸、甘油等中間產物的升高(即高血脂癥),后者作為底物,被運進外周組織,進一步加劇外周組織的TAG合成及高血脂癥.此外,肝臟中的TAG合成增強還能夠通過高水平的乙酰輔酶A等中間產物,變構激活多個糖異生途徑的限速酶[158-159],后者與胰島素抵抗(及其導致的肝糖原分解加強)一起引起高血糖[160].血糖水平的升高又進一步激活SREBP、ChREBP等轉錄因子,加強DNL和TAG堆積[161-162],也可以導致細胞內ROS的增多,進一步導致內質網應激和炎癥反應[163-164].由此可見,上述AMPK對DNL、DAG等脂質合成的抑制以及對β-氧化的加強作用,能夠從根本上緩解包括脂肪、肝臟和肌肉在內的外周組織器官的脂質積累[165-167];其對糖異生的抑制作用、對血液葡萄糖的攝取以及對糖酵解的促進作用,則可以有效降低血糖水平[73-74];而其對炎癥的抑制作用和對細胞內氧化還原穩態的維持,則能夠緩解由此產生的胰島素抵抗[128].上述關于AMPK在肥胖相關代謝性疾病中的作用總結于圖4.
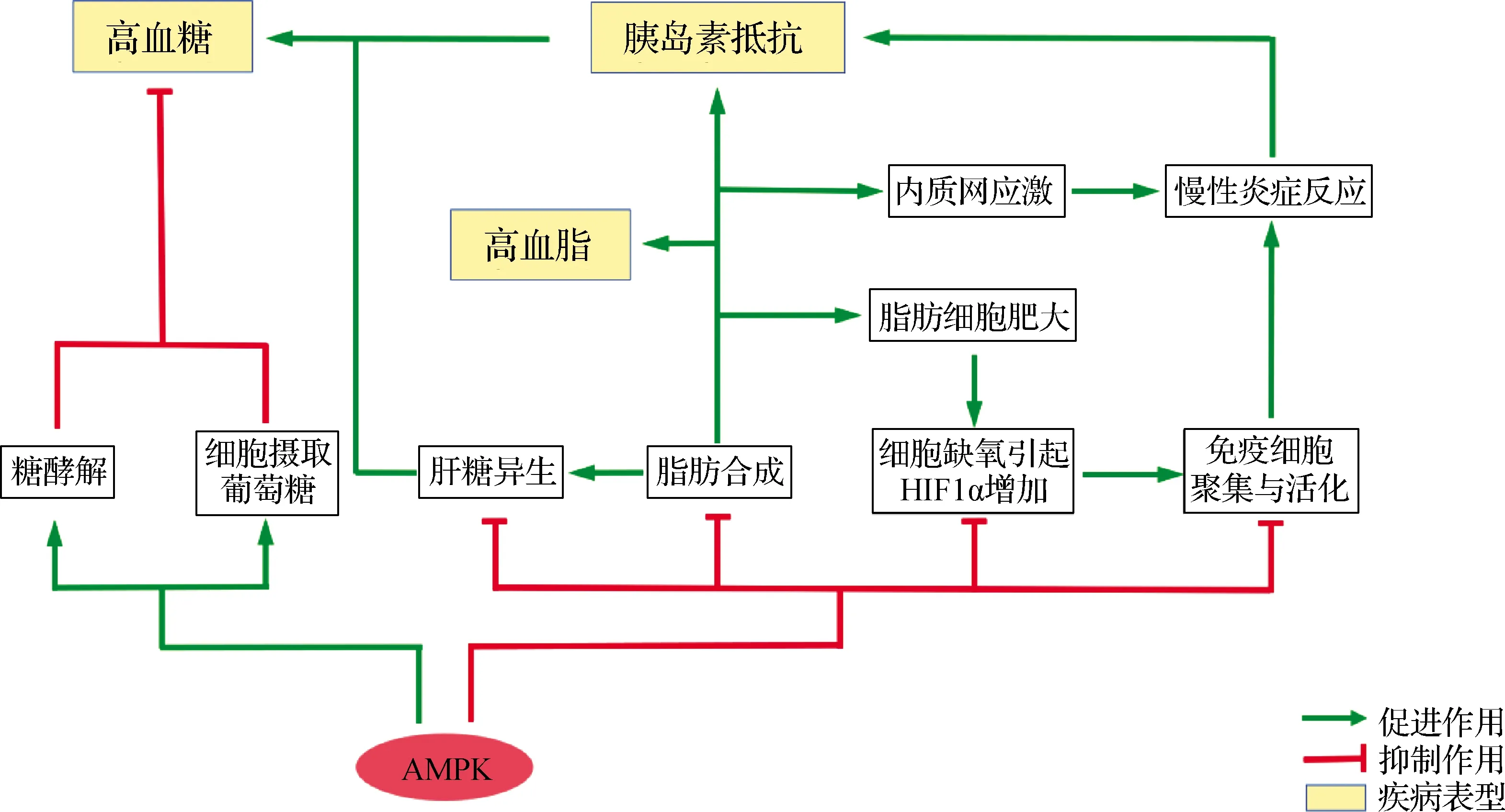
圖4 AMPK在肥胖相關代謝性疾病中的作用
在動物模型和病人中血糖和游離脂肪酸水平的上升以及炎癥反應的加強,能夠普遍抑制AMPK活性[168-170],因此通過藥物重新激活AMPK可起到顯著作用.事實上,以二甲雙胍為代表的AMPK激活劑已經在降低血糖和脂肪肝方面獲得了很好的臨床療效,但其難以進入肌肉、脂肪等組織中發揮激活AMPK的作用[171].因此,合成新的AMPK激活劑日益受到重視,能夠結合并激活AMPK的直接激活劑的研發成為重要方向,如MK-8722[37]、PF-379[167]、A769662[172]、AICAR[173](但需較高劑量[174])、水楊酸/阿司匹林[175]、C2/C13[176]、PT-1[177]和991[178-179]都被證明在緩解肥胖、糖尿病或脂肪肝中發揮作用.此外,TZD[180]、黃連素[181]、白藜蘆醇[182]、人參皂苷[183]、硫辛酸[184]等AMPK的間接激活劑也被證明有類似功效.如前所述,這兩類化合物亟需解決的問題是過強、過廣(無時空特異性)的AMPK激活所引起的副作用.特別地,AMPK已被證明對脂肪肝進一步演變成非酒精性脂肪性肝炎(NASH,是肝纖維化、肝硬化和肝細胞癌發生的前提[185])有明顯抑制作用[186].鑒于目前尚無治療NASH的藥物上市,且前述對脂肪肝有效果的藥物研究也僅局限于動物模型,對相關AMPK激活劑有待進一步探索和改良,使之發揮治療脂肪肝的作用[187].可喜的是,最近有多個來源于天然產物的AMPK激動劑,如雷公藤甲素(triptolide)、β-廣藿香烯(β-patchoulene)和甜菜堿(betaine)表現出強烈的緩解脂肪肝的功能[188-190];而γ-亞麻酸(γ-linolenic acid)能夠通過AMPK調節自噬,抑制脂肪肝引起的肝細胞死亡,后者被認為是脂肪肝發展成為NASH的重要因素[191-193].這些新發現的藥物為脂肪肝和NASH的治療帶來了曙光.
4.2 AMPK與癌癥
近年來,隨著對癌癥病因研究的深入,癌癥作為一種代謝紊亂相關(原因和/或結果)疾病的觀點被逐漸接受[194].鑒于AMPK在抑制合成代謝、阻滯細胞周期等方面的功能,它也成為多種癌癥治療的潛在靶點.遺傳學方面的證據顯示,AMPK的上游激酶LKB1和CaMKKβ都被證明是重要的抑癌基因[195-197],盡管LKB1也能通過ARK抑制癌癥[198].敲除AMPKα1(淋巴細胞中只表達AMPKα1)則會在Eu-myc B淋巴細胞癌模型中加速癌癥的發展[199],類似的效果也在p53缺失的T淋巴細胞癌模型(敲除AMPKβ1)[200]、人第10號染色體缺失的磷酸酶及張力蛋白同源蛋白(PTEN)缺失的急性T淋巴細胞白血病(T-ALL)模型(敲除AMPKα1)和前列腺癌模型(敲除AMPKβ1)中被觀察到[197,201],而MAGE-A3/A6、UBE2O等泛素連接酶介導的AMPKα降解,則在多種模型中促進腫瘤的發生[202-203].除在腫瘤模型水平發揮作用外,AMPK在緩解上述肥胖相關代謝疾病的的同時,也降低了由這些疾病引起的患癌風險[204],且上述AMPK的多種激活劑已被嘗試用于癌癥治療,如:水楊酸和二甲雙胍聯用被證明對前列腺癌和肺癌有抑制作用[205],苯乙雙胍能夠抑制非小細胞肺癌和T-ALL的發展[201,206],而二甲雙胍還可通過AMPK磷酸化程序性死亡配體1(PD-L1)促進其降解,從而加強對乳腺癌和結腸癌的治療效果[207].針對脂質合成代謝對腫瘤細胞生長的關鍵作用[208-209],最近有研究顯示以ACC為靶點設計的抑制劑ND-654對于肝細胞癌的發展有很好的抑制作用[210].此外,AMPK還被報道能夠磷酸化并抑制諸多原癌基因以抑制腫瘤,如:BRAF,經有絲分裂原活化的細胞外信號調節激酶-細胞外調節蛋白激酶(MEK-ERK)途徑[211];YAP,經Hippo途徑[212];GLI1,經Hedgehog途徑[213].
然而需要注意的是,由于腫瘤微環境的多樣性,在許多情況下合成代謝和細胞周期的阻滯反而利于癌細胞的存活和癌癥的發展.當細胞癌變剛完成但處于營養物質匱乏的環境時,AMPK反而有利于腫瘤細胞的存活和癌癥的發展,如:3.2小節所述AMPK對于細胞還原力的維持,有助于腫瘤細胞渡過葡萄糖缺乏的狀態[107,214];AMPK還可通過磷酸化S期激酶相關蛋白2(Skp2)并激活蛋白激酶B(AKT),抵御凋亡并促進乳腺癌細胞抵御包括化療藥物作用在內的多種環境壓力[215],而AMPK對丙酮酸脫氫酶E1亞基(PDHA)的磷酸化和激活,在促進有氧呼吸的同時提升前列腺癌細胞的ATP產率,從而有利于其在遷移過程(處于多種營養缺乏環境,詳見綜述文獻[216])中的存活[217].在小鼠模型上,與胚系敲除AMPK不同,當T-ALL、急性髓性白血病模型中淋巴癌在骨髓處剛發生時便敲除AMPK(此時癌細胞處于葡萄糖水平遠低于血液的環境中),則會促進癌癥的發展[218-219].因此,未來在逐步闡釋癌癥發展的不同階段所經歷的代謝重編程乃至微環境的營養物水平變化基礎上,輔助以時空特異性激活AMPK的用藥策略,將為癌癥的防治帶來極大幫助.
5 AMPK和長壽——長期效應
與對脂肪肝、糖尿病和癌癥等代謝性疾病的短期作用相比,AMPK的長期效應主要體現在它延緩衰老、延長壽命方面的貢獻.
上述AMPK對代謝性疾病的預防作用本身就能夠促進健康與長壽.例如,服用二甲雙胍的糖尿病患者由糖尿病并發癥(包括癌癥)引起的死亡風險明顯降低[220-221],二甲雙胍很可能通過緩解糖尿病的病癥發揮作用[222].AMPK在延長壽命方面的直接作用是通過線蟲(Caenorhabditiselegans)、果蠅(Drosophilamelanogaster)等模式生物的相關研究發現的,在線蟲中表達有活性的AMPK,能夠顯著延長其壽命[223-224];在果蠅中,AMPK和LKB1的表達都可以延長其壽命[225-226].此外,多種已知可促進健康長壽的生活方式也能夠通過AMPK發揮作用.例如,目前唯一在所有已驗證過的動物(包括非人靈長類和人類[227-228])中都能延長壽命、提高生活質量的生活方式——卡路里限制(CR),被發現能激活AMPK[229-230],且在線蟲和果蠅中已證明缺失AMPK可影響CR介導的延長壽命的效果[224-225].上述AMPK激活劑如二甲雙胍和白藜蘆醇,在延長壽命、延緩衰老方面的功能也已得到廣泛研究:兩者都能夠顯著延長線蟲[231-232]、果蠅[232-233]和小鼠[182,234]的壽命,且在線蟲和小鼠中均呈現AMPK依賴性[231,235-237].二甲雙胍和白藜蘆醇的上述作用模擬了CR的效果,因而被稱為CR模擬物(CRM),目前,AMPK是設計CRM的核心靶點之一[238].鑒于二甲雙胍廣泛的臨床應用(白藜蘆醇的成藥性仍需進一步改善[239])和已被反復證實的安全性,加之其對非糖尿病人群的患癌率有明顯抑制作用[240],它在健康人群中抗衰老作用的臨床研究已經開始[241].
從機制上看,AMPK的多個下游底物都和壽命有關.目前發現和壽命相關的主要信號通路有3類:Sirtuins的活化、mTORC1的抑制以及胰島素和胰島素類生長因子途徑的抑制[242-244].上述AMPK對Sirtuins的調節[245]、對TSC和Raptor等分子的磷酸化[246]以及對CRTCs和FOXO的抑制[224,247],則分別介導了這3個過程.在細胞層面,上述AMPK在維持線粒體的數量與質量[248-249]、mitohormesis[250]、自噬作用[251-252]、蛋白合成[253]等方面的功能也被證明與壽命密切相關.類似于肥胖和糖尿病,研究表明衰老也是一種機體的慢性炎癥反應,被稱為“inflammaging”[254],而AMPK對免疫細胞炎癥化的抑制則能延緩衰老的過程[255].細胞的衰老,特別是免疫細胞和干細胞的衰老,被認為和壽命有極大關聯[256-257].盡管有研究表明細胞衰老可能有抑制癌癥發生的功效[258],但衰老細胞裂解對于延長壽命、延緩衰老有重要作用[259-260].AMPK對于細胞周期的調節作用已被證明能夠抑制衰老[261-262].而目前引發衰老細胞裂解最有效的化合物Fisetin[263]也被證明能激活AMPK[264].此外,生物體的節律維持對于衰老和壽命有重要影響[265],而AMPK已被證明在該過程中起關鍵作用[266].最后,需要特別注意的是,盡管AMPK與健康長壽之間有天然而密切的聯系,但兩者之間的因果關系,特別是AMPK在引起長壽的不同策略下的具體分子機制及其與長壽的直接聯系,尚未完全闡明.例如:和直接表達有活性的AMPK不同,二甲雙胍對線蟲壽命的延長不依賴于FOXO,而依賴于NRF所介導的類似于mitohormesis提高有氧呼吸效率的機制[224,231];同樣是二甲雙胍,作用于線蟲的老年期,則反而會破壞線粒體功能并引起壽命的縮短[267].這暗示AMPK對長壽的調節處于復雜的時空網絡下,在“正確的時間”和“正確的地點”做“正確的事”,而進一步詳細闡明其中的機制將為延長壽命、維持健康提供更多思路.
6 總結與展望
AMPK作為一種調控細胞能量代謝的重要分子,通過對能量、葡萄糖等的感知維持代謝穩態,并在多種代謝綜合征乃至衰老中行使重要的功能,為人類最終戰勝代謝性疾病帶來了信心,也因此成為重要的藥物靶點.然而如本文所述,由于AMPK激活和功能的時空網絡的復雜性,靶向AMPK治療代謝性疾病的策略尚存在一定困難.今后,圍繞著AMPK調節和功能上的“時空性”開展日益深入的研究,將多維度展示這一代謝性疾病治療的靶標作用機制和功能,為最終通過操控AMPK以促進人類的健康長壽做出貢獻.
致謝:感謝導師林圣彩院士和為AMPK溶酶體途徑的發現做出貢獻的所有前輩和后輩,還要感謝廈門大學生命科學學院的培養,并感謝廈門大學《生物學實踐》課程和參與此課程的“追糖溯源”實踐隊全體成員為本文最終成形做出的貢獻.
猜你喜歡
美與時代·美術學刊(2022年3期)2022-04-27 01:18:15
火花(2019年12期)2019-12-26 01:00:28
人大建設(2019年6期)2019-10-08 08:55:48
人大建設(2019年12期)2019-05-21 02:55:32
雜文月刊(2018年21期)2019-01-05 05:55:28
人大建設(2017年6期)2017-09-26 11:50:44
學苑創造·A版(2015年11期)2016-01-14 09:03:27
俄羅斯問題研究(2012年1期)2012-03-25 09:54:45
中國火炬(2010年12期)2010-07-25 13:26:22
中國火炬(2010年8期)2010-07-25 11:34:30
