UPLC-MS/MS 測定茅蒼術中蒼術素和蒼術苷的含量
2022-08-10 05:47:28張文文吳琴燕殷錫峰倪衛東梁紅芳
江西農業學報 2022年5期
張文文,吳琴燕,殷錫峰,孟 嫻,倪衛東,梁紅芳
(1.鎮江市農產品質量檢驗測試中心,江蘇 鎮江 212009;2.江蘇丘陵地區鎮江農業科學研究所,江蘇 句容 212400;3.句容市中醫院,江蘇 句容 212400)
蒼術為菊科多年生草本植物,根莖為中藥蒼術的基原植物之一,具有燥濕健脾,祛風散寒,明目的功效[1]。蒼術屬植物在我國分布較廣,因江蘇茅山地區的蒼術品質較好,故名為茅蒼術[2]。蒼術中主要藥用成分為蒼術素、蒼術苷、β-桉葉醇、白術內酯Ⅰ、白術內酯Ⅱ和白術內酯Ⅲ等[3]。2015年版《中國藥典》將蒼術素規定為蒼術的指標性成分,含量不低于0.3%[3],而在茅蒼術中該含量約為0.6%[4]。大量報道表明,蒼術苷存在于蒼耳子中,含量達到0.1%[5],蒼術苷是否存在于蒼術中暫未見報道。目前,對蒼術化學成分的檢測主要采用HPLC法和GC-MS法進行測定,存在樣品基質干擾較大、前處理復雜、檢測時間長等問題[3],本研究擬建立茅蒼術中蒼術素和蒼術苷的UPLC-MS/MS檢測方法,以期為茅蒼術的質量評價提供科學依據。
1 實驗材料與方法
1.1 儀器、試劑和材料
超高效液相色譜:Agilent 1290;質譜檢測器:ABsciex 4500;電子天平:XP105DR 型,賽多利斯科學儀器有限公司;超純水系統:AWL-020I-P,艾科浦儀器有限公司;超聲波清洗器:KQ-250E型,昆山禾創超聲儀器有限公司;醫用離心機:H2050R 型,湖南湘儀實驗室儀器開發有限公司。
甲醇和乙腈(色譜純,美國默克公司);乙酸銨(≥98%,德國CNW 公司);蒼術素、蒼術苷標準樣品購買于成都曼思特生物科技有限公司,純度>98%。質譜用水為屈臣氏蒸餾水。
1.2 標準溶液配置
母液配置:分別稱取蒼術素和蒼術苷1 mg,蒼術素加入1 mL丙酮溶解,蒼術苷加入1 mL甲醇溶解,分別配置成1 mg/mL的母液,-20 ℃保存后,備用。
標準溶液的配置:按照梯度稀釋法,分別配置蒼術素濃度為100、50、25、12.5、6.125 mg/L的單一標準品溶液,蒼術苷濃度為4、2、1、0.5、0.25、0.125 mg/L的單一標準品溶液。
1.3 儀器工作條件
色譜柱:Agilent C 18色譜柱(100 mm×2.1 mm,1.7 μm);流動相:0.1%甲酸混合水溶液(A)和0.1%甲酸混合乙腈溶液(B),洗脫梯度:0~1 min,40%A;1~5 min,40%~10%A;5~9 min,10%A;9.00~9.10 min,10%~40%A;9.10~12.00 min,40%A。進樣體積為1 μL;流速為0.2 mL/min;柱溫為40 ℃。
質譜條件:離子源為電噴霧離子源(ESI);離子源加熱溫度(TEM)450 ℃;離子源電壓(IS)為-4500 V;碰撞氣(CAD)為medium;氣簾氣(CUR)為10;霧化氣(GS1)為45 ℃;輔助氣(GS2)為45℃。檢測方式為多反應檢測模式(MRM),監測離子及相應參數見表1所示。
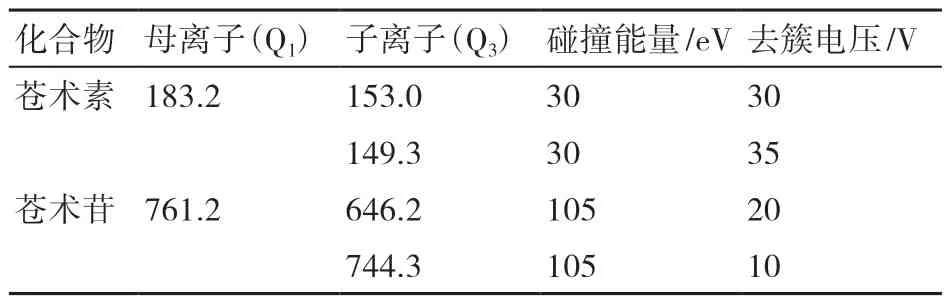
表1 MRM監測模式條件下質譜參數
1.4 樣品前處理
茅蒼術(購買于句容茅山農戶)通過試驗粉碎機(FW100型,天津市泰斯特儀器有限公司)對蒼術進行粗粉碎至過40目篩網后備用。
提取溶劑試驗:準確稱取1 g蒼術粉末,分別采用水、乙腈、丙酮、異丙醇、乙醚、乙酸乙酯和甲醇7種提取劑40 mL,室溫條件下超聲(250 W,頻率35 kHz)提取30 min,離心取上清待測。
提取時間試驗:準確稱取1 g蒼術粉末,選擇合適的提取溶劑40 mL,在室溫條件下,分別超聲提取5、15、30、45、60 min,離心取上清待測。
提取甲醇含量試驗:準確稱取1 g蒼術粉末,分別取40 mL濃度為0、20%、40%、60%、80%、100%的甲醇,室溫條件下超聲提取30 min,離心取上清待測。
提取液固比試驗:取40 mL甲醇和一定量的蒼術粉,在液固比分別為5、10、20、40、80和120 mL/g時,室溫條件下超聲提取30 min,離心取上清待測。
2 結果與分析
2.1 儀器條件優化
2.1.1 質譜條件優化 依據每個化合物響應值的差別,把蒼術素和蒼術苷分別配置為25.0和0.5 mg/L的標準品溶液,進行質譜條件優化,2種化合物均在正離子掃描模式下,蒼術素獲得了準分子離子[M+H]+,蒼術苷獲得的脫鉀離子[M-K+H]+,響應明顯較高,以此作為母離子,進行子離子掃描分析,圖1、圖2為2種化合物的二級掃描質譜圖,選擇響應值高的離子為二級特征離子,該離子碎片與文獻報道結果一致[6-7]。根據特征離子,對去簇電壓、碰撞能量等參數進行優化,使得離子對的信號達到最高,優化后的質譜條件如表1所示。
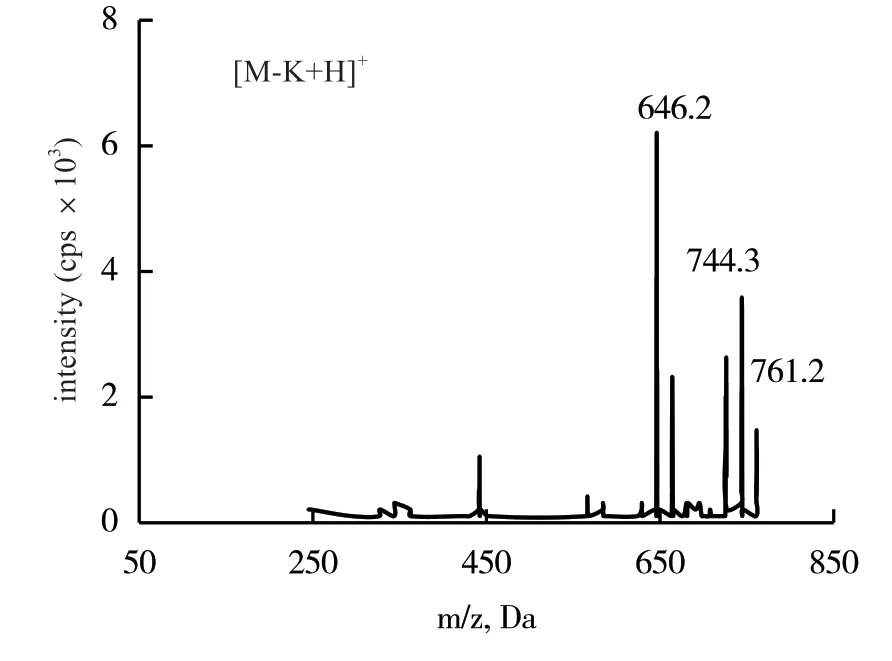
圖1 蒼術苷二級掃描質譜圖
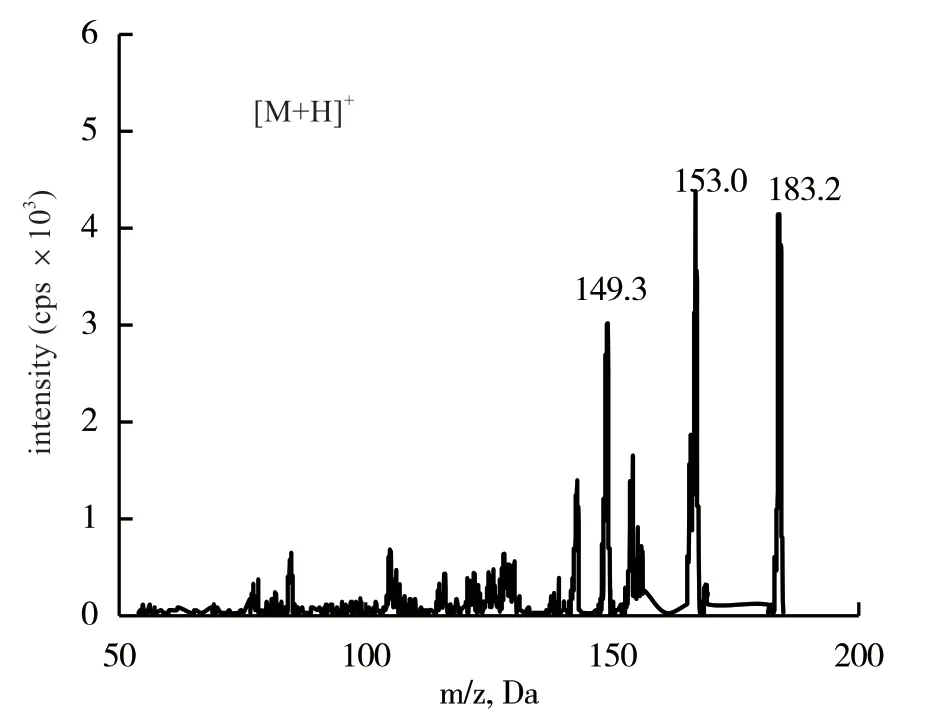
圖2 蒼術素二級掃描質譜圖
2.1.2 色譜條件優化 根據2種化合物的理化性質,選用C18色譜柱,選擇5 mol/L乙酸銨(含0.1%甲酸)為水相,乙腈為有機相,經過反復實驗確定1.2所述洗脫程序,可以在8 min內獲得良好的分離效果和適當的出峰時間。蒼術素在4.5 min左右出峰,蒼術苷在1.5 min左右出峰,且基質中雜峰較多,此梯度可以實現蒼術素與雜質較好的分離(圖3、圖4)。蒼術苷分子量較大、干擾小,樣品中含量低。
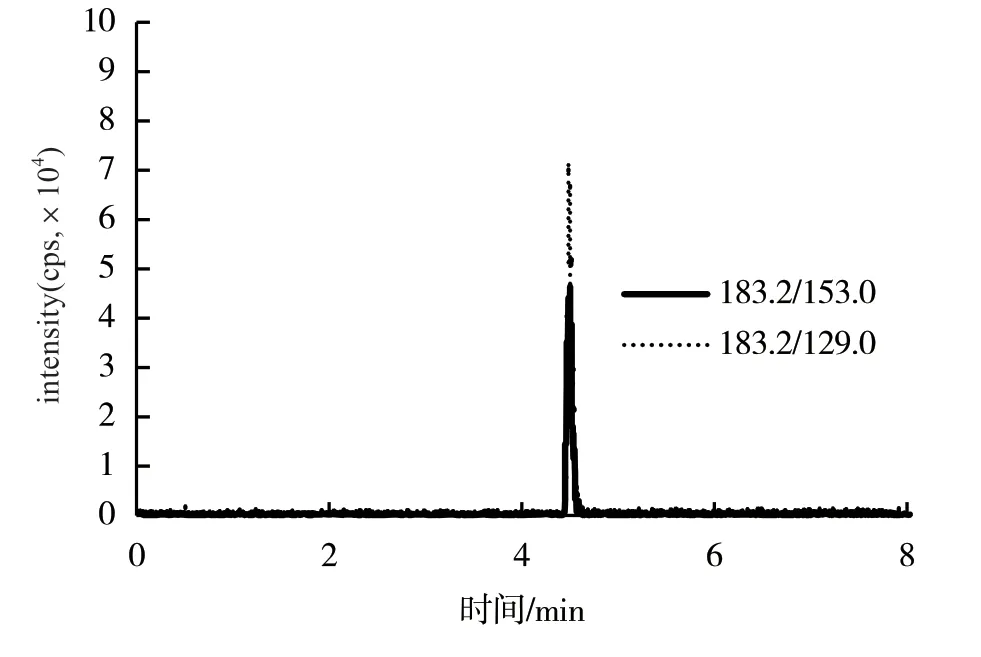
圖3 標樣中蒼術素色譜圖

圖4 標樣中蒼術苷色譜圖
2.2 線性關系、檢出限和定量限
根據1.2標準溶液配制濃度數據,獲得方法的線性關系、檢出限和定量限如表2所示。

表2 線性關系、檢出限和定量限
2.3 樣品前處理條件優化
2.3.1 提取溶劑的影響 乙腈[8]、丙酮[8]、異丙醇[9]、乙醚[9]、乙酸乙酯[10]、甲醇[11]和三氯甲烷[8]是非極性物質的主要提取劑,蒼術素為非極性物質,蒼術苷為極性物質,采用水、乙腈、丙酮、異丙醇、乙醚、乙酸乙酯、甲醇和三氯甲烷的同時提取蒼術素和蒼術苷(圖5),發現采用甲醇提取,蒼術素和蒼術苷提取量均高于其他提取劑,分別為0.908 mg/g和7.010 μg/g。
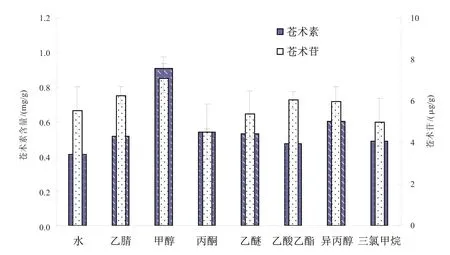
圖5 不同提取溶劑對蒼術素和蒼術苷含量的影響
2.3.2 提取時間的影響 由圖6可知,提取時間對蒼術素有一定的影響,隨著時間的延長,提取量升高,30 min后不再變化;提取時間對蒼術苷無明顯影響,30 min為最佳提取時間。

圖6 提取時間對蒼術素和蒼術苷含量的影響
2.3.3 甲醇濃度的影響 據文獻報道,適量的水對原料具有溶脹作用,可以促進有效成分的溶出。由圖7可知,在對倉術的提取過程中,甲醇濃度的不同,對蒼術苷的影響較大,濃度越高,提取量越高;而對蒼術素的影響不明顯,因此,水的溶脹作用并未有效促進蒼術苷和蒼術素的提取量,最佳提取溶劑為100%濃度甲醇。
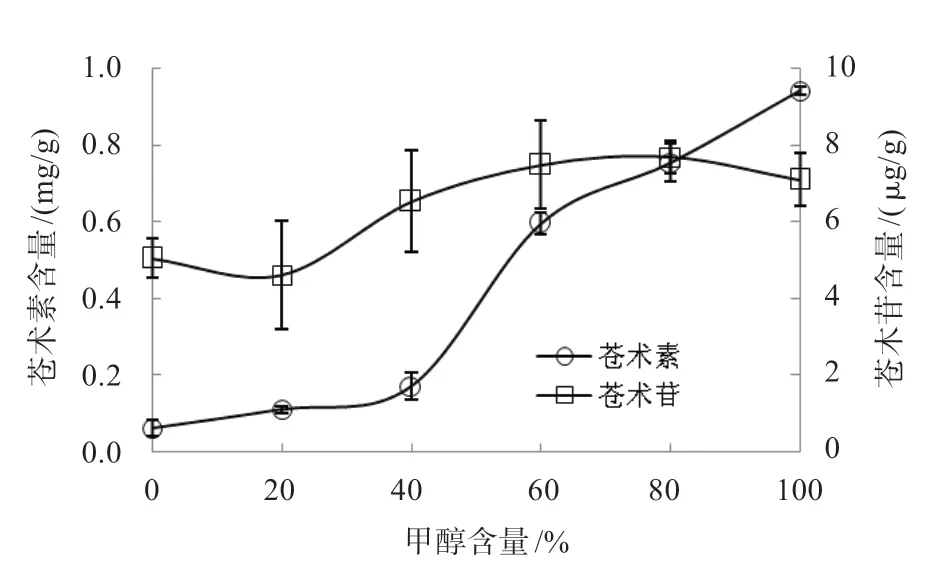
圖7 甲醇濃度對蒼術素和蒼術苷含量的影響
2.3.4 提取液固比的影響 液固比的影響如圖8所示,由圖8可知,液固比在80 mL/g時,蒼術素和蒼術苷提取量達到最佳值,繼續增加液固比,含量變化不明顯。
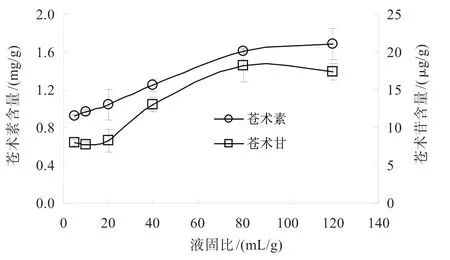
圖8 液固比對蒼術素和蒼術苷含量的影響
2.4 回收率和標準偏差
取40 mL甲醇,加入0.5 g蒼術粉,并加入一定量的蒼術素和蒼術苷標品,使蒼術中分別含有1.0、0.5、0.2 mg/g的蒼術素標樣和2.5、10.0和20.0 μg/g的蒼術苷標樣,每個濃度重復4次,計算該方法的回收率和相對標準偏差(RSD),結果表明,3個濃度加標水平下,蒼術素和蒼術苷的回收率分別為77.9%~95.0%和72.5%~106.6%,相對標準偏差為5.3%~9.5%和9.5%~12.6%,均符合檢測要求(表3)。

表3 樣品加標回收率和相對標準偏差
3 結論
建立了使用LC-MS/MS快速測定茅蒼術中蒼術素和蒼術苷的方法,通過優化質譜和色譜參數,2種化合物均具有較高的離子化效應的離子對和最佳檢測條件。采用超聲提取,對提取溶劑、提取時間、甲醇濃度和固液比4個提取條件進行優化,發現以甲醇為提取劑,在液固比為80 mL/g,超聲提取30 min,蒼術素和蒼術苷的提取量均達到了較高水平,分別約為1.6 mg/g和18 μg/g。該方法前處理簡單、分析快速、定性定量準確、檢測限低,可以滿足茅蒼術中對蒼術素和蒼術苷的檢測要求。
