基于單樣本基因富集分析分型構建肺腺癌的風險預測模型
2022-09-14 11:46:24高晨吳林玉孔寧婁新璟郭勇許茂盛
浙江醫學 2022年16期
高晨 吳林玉 孔寧 婁新璟 郭勇 許茂盛
肺癌是世界上發病率第二及死亡率最高的惡性腫瘤[1]。據研究報道,中國肺癌患者的5年總生存期僅為19.8%[2]。盡管手術方式、靶向治療等技術的發展明顯改善了肺癌患者的預后,但是仍然達不到預期效果[3-4]。肺腺癌是肺癌最常見的病理亞型,目前判斷肺腺癌預后的主要依據是腫瘤的TNM分期,但這種方法存在較大的差異,精確性也有待提高[5-6]。多種分子機制參與了肺腺癌的發生、發展,只有對這些機制進行更深入的研究,才能更好地預測患者的預后,指導臨床治療,提高患者總生存期[7-8]。因此,臨床上迫切需要尋找更為精準預測肺腺癌患者預后的分子生物標志物。本研究擬通過基于單樣本基因富集分析(single sample gene set enrichment analysis,ssGSEA)的方法對來自癌癥基因組圖譜(The Cancer Genome Atlas,TCGA)數據庫及基因表達數據庫(Gene Expression Omnibus,GEO)中肺腺癌患者轉錄組的數據進行系統性生物信息學分析并構建肺腺癌的風險預測模型,為臨床醫師判斷肺腺癌患者總生存期提供輔助工具,同時為尋找潛在的靶向治療藥物提供參考依據。
1 資料和方法
1.1 數據獲取及整理 從基因組數據共享(Genomic Data Commons,GDC)官方網站(https://portal.gdc.cancer.gov)以及GEO官方網站(https://www.ncbi.nlm.nih.gov/geo)獲取TCGA數據庫及GSE26939數據集肺腺癌患者的轉錄組數據以及相關患者臨床數據。TCGA肺腺癌數據庫有515例患者的594個樣本,其中59個為正常樣本/癌旁樣本,535個為腫瘤樣本。GEO數據集作為外部驗證集,數據集中有116例肺腺癌患者的116個腫瘤樣本。從免疫學數據庫和分析門戶(The Immunology Database and Analysis Portal,ImmPort)網站(https://www.immport.org/home)共下載免疫相關的基因1 793個。從順反組數據瀏覽器(Cistrome Data Browser,Cistrome DB)轉錄因子網站(http://www.cistrome.org)共下載轉錄因子318個。
1.2 ssGSEA及免疫分析 對來自TCGA數據庫肺腺癌患者的轉錄組數據,運用ssGSEA分析及聚類分析,從而獲得樣本的免疫評分及不同免疫評分組;運用ESTIMATE分析,獲得腫瘤樣本的腫瘤微環境分數,并進一步分析腫瘤微環境分數在不同免疫評分組中的差異。運用CIBERSORT算法計算樣本中22種腫瘤浸潤免疫細胞的比例,并分析不同免疫評分組中腫瘤浸潤免疫細胞比例的差異。在GSEA軟件(版本4.1.0)上基于不同免疫評分組進行GSEA富集分析,獲得顯著的富集通路,并分析患者腫瘤樣本的基因表達量在不同免疫評分組中的差異,獲得在不同免疫分型組中的差異表達基因。通過取差異表達基因列表與免疫相關基因列表的交集,獲得存在差異的免疫基因。
1.3 預后基因的篩選 在得到TCGA與GEO數據集中差異免疫基因表達量后,對其進行批次矯正。同時對患者進行進一步篩選,納入標準:(1)病理檢查確定為肺腺癌;(2)具有可獲取的轉錄組數據;(3)具有可獲取的臨床數據。排除標準:(1)未同時具有可獲取的轉錄組數據及臨床數據;(2)缺少相關生存數據。最終確定納入下一步研究的肺腺癌患者,其中TCGA數據庫504例,GEO數據庫115例。將來自TCGA數據集中的患者以7∶3隨機分為訓練集356例和內部驗證集148例。同時將GEO數據集的患者作為外部驗證集。基于訓練集的數據,通過單因素Cox回歸分析,篩選出肺腺癌患者預后相關的差異免疫基因。將預后相關的差異免疫基因與轉錄因子進行相關性分析,以及在Cytoscape軟件(版本3.8.2)上進行相關蛋白互作網絡的分析。
1.4 預后模型的建立 通過套索算法(least absolute shrinkage and selection operator,Lasso)回歸對篩選出的肺腺癌患者預后相關差異免疫基因進行降維,并利用多元逐步Cox回歸模型篩選出最優的差異免疫基因集合構建肺腺癌的風險預測模型,并獲得每個樣本的風險分數(Riskscore)。基于獲得Riskscore的中位數,將患者分為高風險組及低風險組。運用內部及外部驗證集的數據對預測模型進行檢驗,并采用ROC曲線及校正曲線來顯示預測模型在訓練組和驗證組中預測模型的效能。采用Kaplan-Meier法對訓練集、內部驗證集、外部驗證集進行生存分析。
1.5 聯合模型及列線圖構建 運用單因素及多因素Cox風險回歸,將上述獲得的Riskscore與患者的臨床特征(性別、年齡、腫瘤分期)進行獨立預后分析,獲得肺腺癌患者獨立預后因子并構建聯合模型。采用ROC曲線、校正曲線及列線圖分析該聯合模型的效能及臨床實用性。
1.6 統計學處理 采用R 4.0.5統計軟件。運用的R語言包有 GSVA、limma、GSEABase、sparcl、Rtsne、ggplot2、estimate、pheatmap、reshape、reshape2、ggpubr、preprocessCore、venn、sva、survival、ggalluvial、dplyr、caret、glmnet、survminer、timeROC、rms、ggExtra、tidyverse、regplot。差異分析使用Wilcoxon秩和檢驗,相關性分析采用Pearson相關。P<0.05為差異有統計學意義。
2 結果
2.1 免疫評分及相關差異分析 根據腫瘤樣本的ssGSEA免疫評分及聚類分析結果,將535個腫瘤樣本分為高免疫評分組313個和低免疫評分組222個。高免疫評分組、低免疫評分組與腫瘤微環境分數的相關性見圖1a(插頁),其中高免疫評分組中的腫瘤純度較低免疫評分組低,基質分數、免疫分數及ESTIMATE分數較低免疫評分組高,見圖1b(插頁)。運用Wilcoxon秩和檢驗分析基于CIBERSORT算法的12種腫瘤浸潤免疫細胞的比例在高免疫評分組和低免疫評分組中的差異有統計學意義,見圖1c(插頁)。在高免疫評分組中顯著富集通路有“cytokine-cytokine receptor interaction”“natural killer cell mediated cytotoxicity”“cell adhesion molecules”“T cell receptor signaling pathway”和“chemokine signaling pathway”等,見圖2a(插頁)。根據差異分析,在高免疫評分組和低免疫評分組中的差異表達基因有1 447個,見圖2b(插頁)。將差異表達基因與從ImmPort網站獲取的免疫相關基因取交集后,獲得免疫相關的差異表達基因有382個,見圖2c(插頁)。
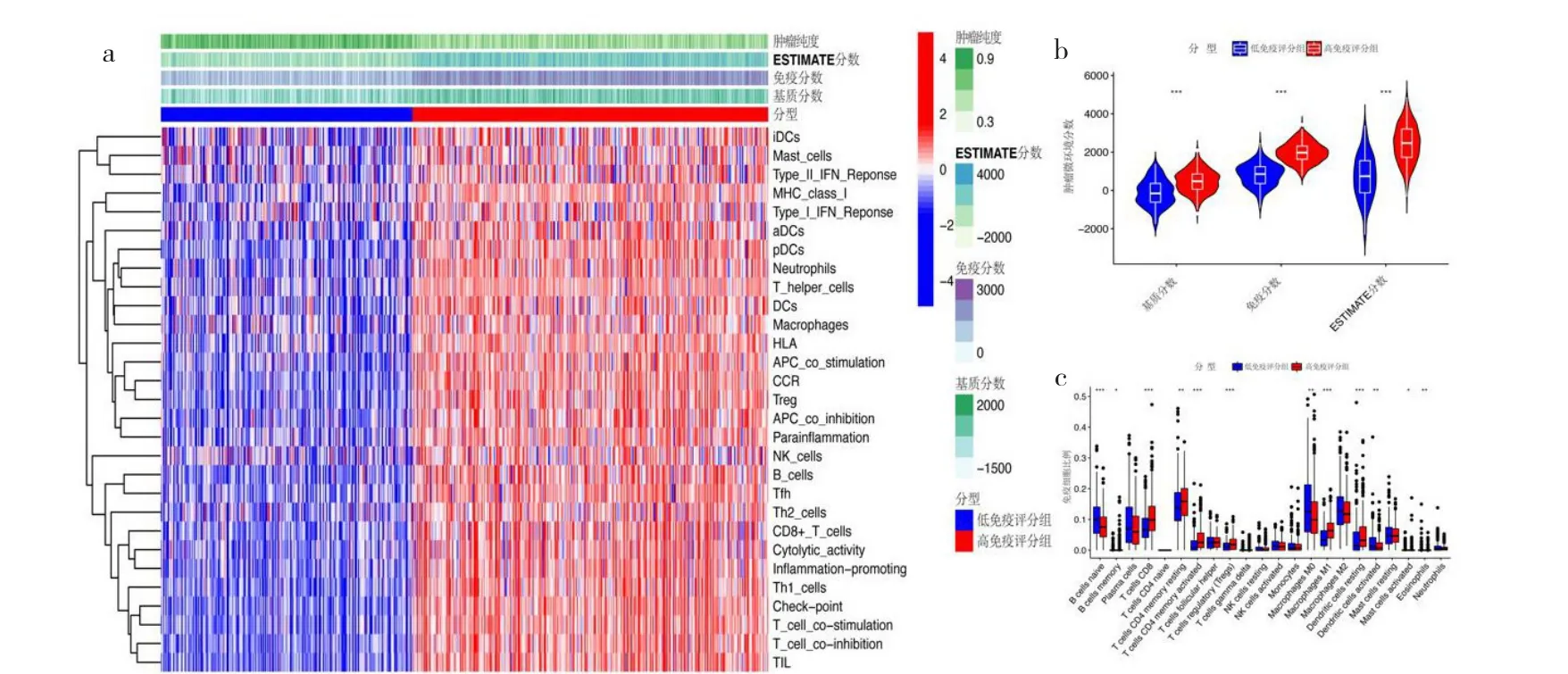
圖1 肺腺癌患者免疫分析及不同免疫評分組的差異分析(a:腫瘤純度、ESTIMATE分數、免疫分數及基質分數與高免疫評分組和低免疫評分組之間的相關性,高免疫評分組中腫瘤純度較低免疫評分組低,基質分數、免疫分數及ESTIMATE分數較低免疫評分組高;b:基質分數、免疫分數及ESTIMATE分數在高免疫評分組和低免疫評分組間差異有統計學意義,高免疫評分組的3個分數均比低免疫評分組高;c:基于CIBERSORT算法的12種腫瘤浸潤免疫細胞的比例在高免疫評分組和低免疫評分組中差異有統計學意義)
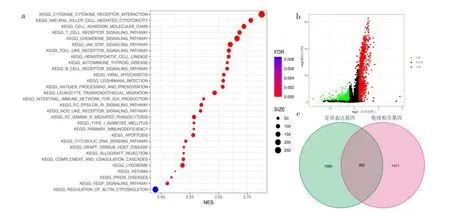
圖2 GSEA富集分析及差異免疫基因確定(a:高免疫評分組中前5個顯著富集通路有“cytokine-cytokine receptor interaction”“natural killer cell mediated cytotoxicity”“cell adhesion molecules”“ T cell receptor signaling pathway”和“chemokine signaling pathway”;b:火山圖顯示了在高免疫評分組和低免疫評分組中差異表達基因,綠色的是在高免疫評分組中低表達的基因,紅色的是在高免疫評分組中高表達的基因;c:韋恩圖顯示了綠色的是1 447個差異表達基因,粉紅色的免疫基因是1 793個,交集后免疫相關的差異表達基因有382個)
2.2 確定預后相關的差異免疫基因及其相關性分析 在對TCGA數據集及GEO數據集的轉錄組數據進行批次矯正且將其與上述免疫相關的差異表達基因列表取交集后,共得到219個差異表達基因的轉錄組數據。經過進一步篩選,共納入619例肺腺癌患者,男287例,女332例,年齡33~90(65.04±10.20)歲,患者基線資料見表1。在訓練集中,采用單因素Cox風險回歸模型對219個基因的轉錄組數據及生存信息進行分析后,得到31個預后相關的差異免疫基因,見圖3a(插頁),這些預后相關的差異免疫基因與轉錄因子的相關性以及蛋白互作網絡關系見圖3b、c(插頁)。

表1 TCGA及GEO數據集中肺腺癌患者臨床資料
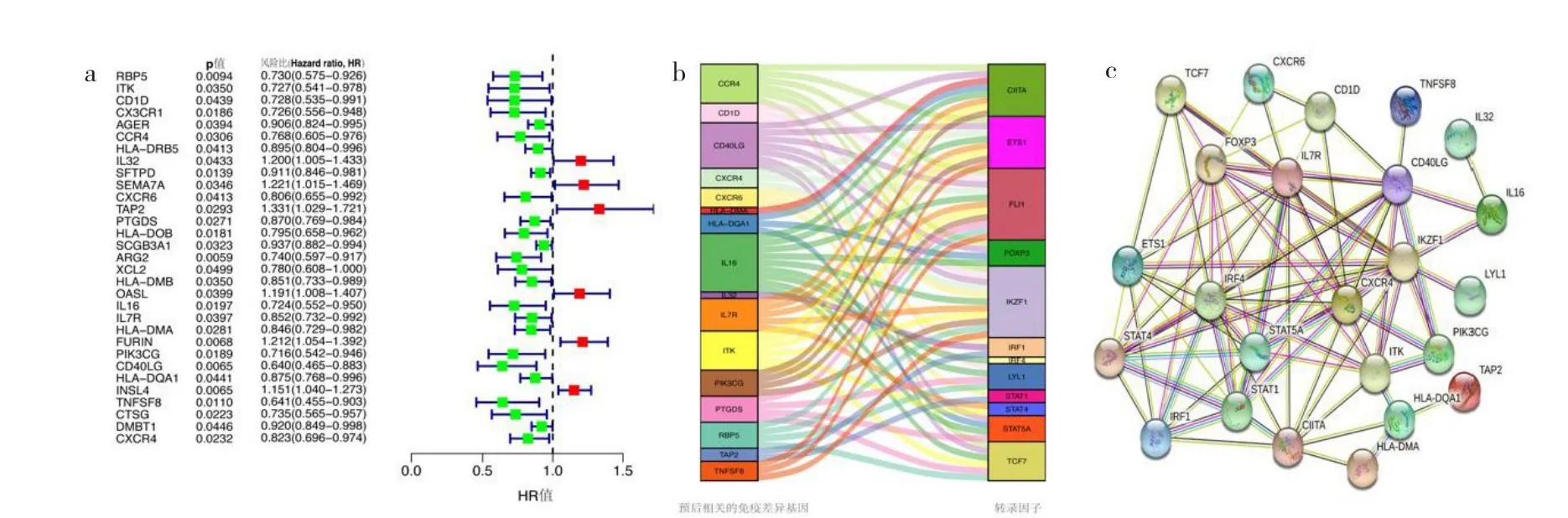
圖3 預后相關的差異免疫基因及與轉錄因子相關性分析[a:森林圖顯示基于單因素Cox風險回歸分析獲得的31個預后相關的差異免疫基因;b和c:桑基圖(b)顯示與轉錄因子具有顯著相關性的16個預后相關差異免疫基因(相關系數≥0.5,P<0.01),PPI圖(c)顯示了其蛋白互作的關系]
2.3 預后模型的構建及生存分析 經過Lasso回歸及多元逐步Cox風險回歸分析降維后,得到最優的差異免疫基因數據集并建立風險預測模型以及得到Riskscore,并根據訓練集數據Riskscore的中位數,將訓練集、內部驗證集及外部驗證集的患者分為高風險組及低風險組,其中訓練集高風險組178例,低風險組178例;內部驗證集高風險組76例,低風險組72例;外部驗證集高風險組50例,低風險組65例。該模型的公式如下:Riskscore=EXP[(-0.211 434 889)×CX3CR1+(0.293 765 44)×IL-32+(-0.071 165 091)×SFTPD+(-0.333 423 936)×CXCR6+0.419 839 844×TAP2+(-0.269 368 749)×HLA-DOB+(-0.374 908 714)×ARG2+0.178 859 695×FURIN+(-1.040 515 441 267 49)]。該風險預測模型在對訓練集及兩個驗證集患者的生存預測上均具有較好的表現,預測患者5年總生存期的AUC分別為0.703、0.713、0.750,見圖4a-c(插頁)。同時校正曲線分析顯示該模型預測的1、3、5年患者總生存期都與實際總生存期較為一致,見圖4d-f(插頁)。此外,高風險組和低分險組患者的總生存期在3個數據集中差異均有統計學意義,見圖4g-i(插頁)。
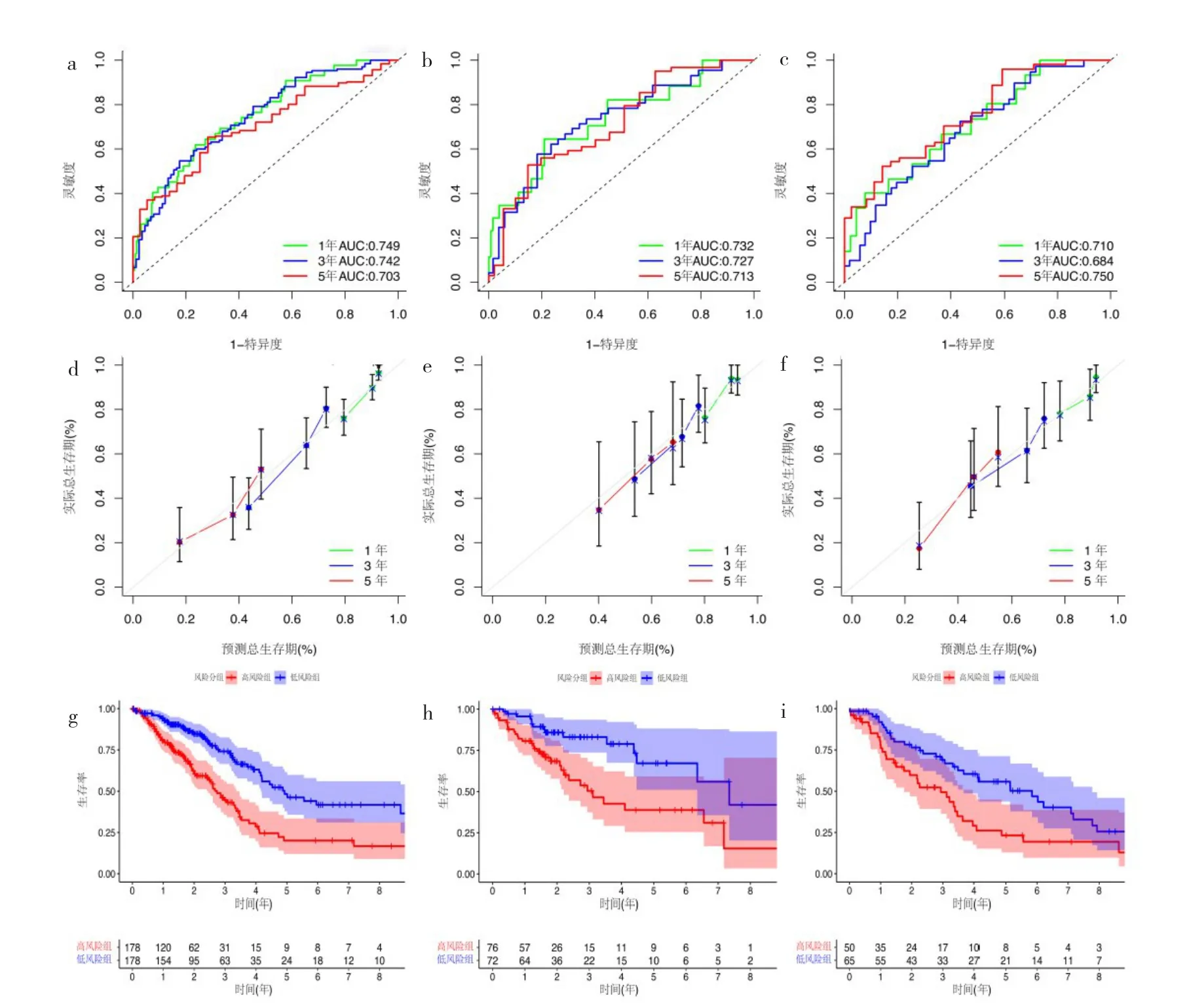
圖4 預測模型的ROC曲線、校正曲線分析及生存分析[a-c:訓練集、內部驗證集、外部驗證集的預測模型ROC曲線;d-f:訓練集、內部驗證集、外部驗證集的校正曲線分析,結果顯示預測結果與實際結果較為一致;g-i:在訓練集、內部驗證集、外部驗證集中,高風險組和低風險組患者生存曲線差異均有統計學意義(均P<0.01),低風險組的患者總生存期較高風險組高]
2.4 獨立預后分析及列線圖的構建 經單因素及多因素Cox風險回歸的分析結果顯示,肺腺癌的分期及預測模型的Riskscore可作為肺腺癌患者的兩個獨立預后因子,見表2。肺腺癌的分期聯合預測模型的Riskscore構建聯合模型的1、3、5年總生存期的AUC分別為0.789、0.763、0.746。校正曲線分析顯示該模型預測的1、3、5年患者總生存期與實際總生存期較為一致。同時該聯合模型臨床應用的列線圖見圖5。
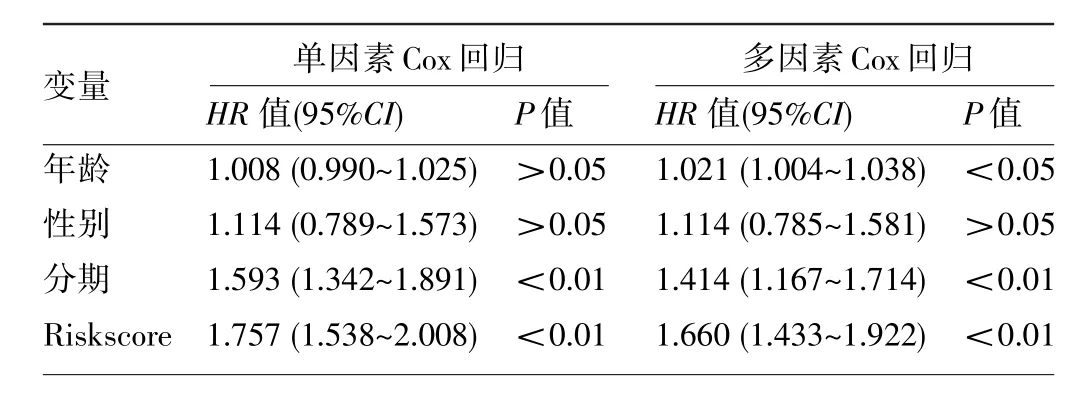
表2 單因素及多因素Cox回歸分析
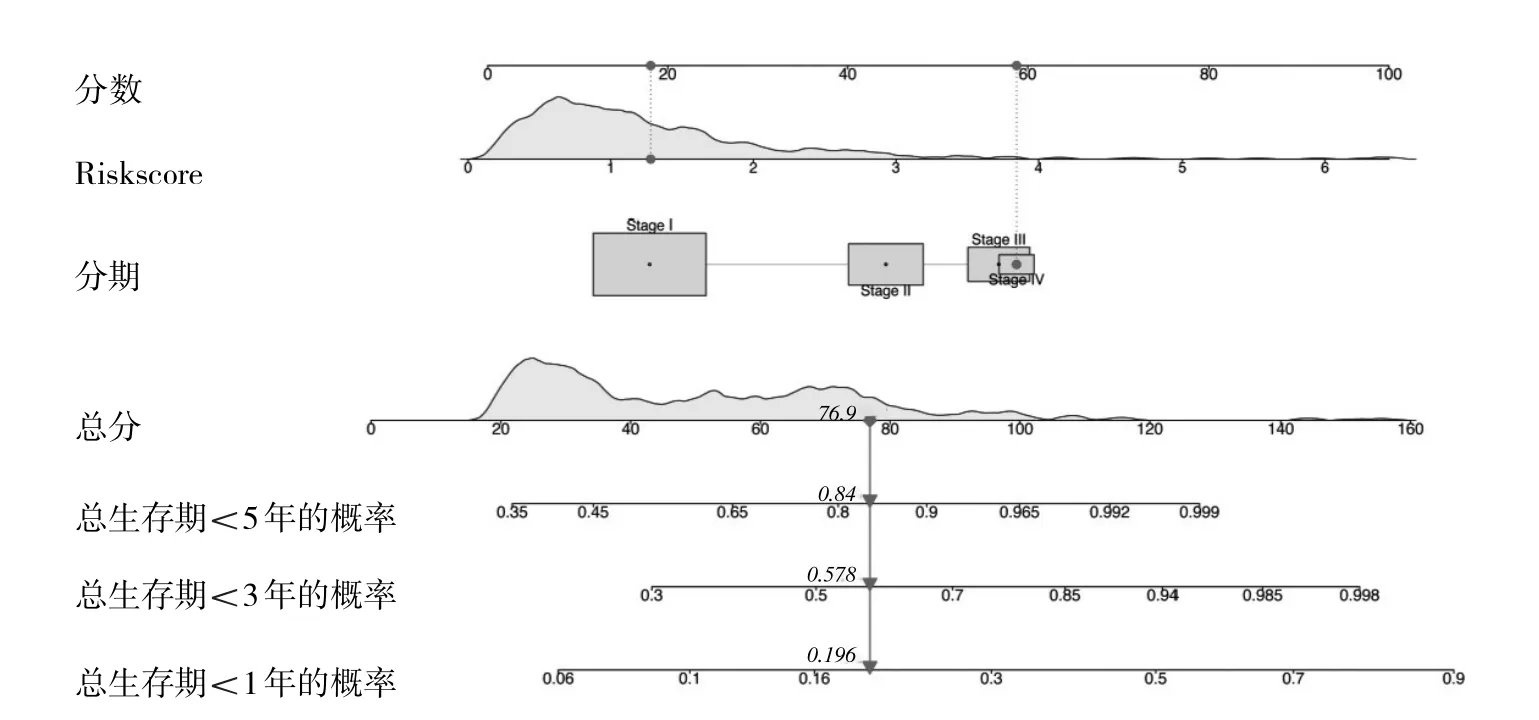
圖5 聯合模型的列線圖的構建(點表示1例患者,其Riskscore與分期Ⅳ期的總得分為76.9分,總生存期<1年的概率是0.196,<3年的概率是0.578,<5年的概率0.84)
3 討論
ssGSEA是能對單個樣本的通路富集情況進行量化并評分的分析方法,例如免疫相關通路等[9]。同時已有不少研究報道了腫瘤微環境中免疫細胞浸潤水平與腫瘤的發生、發展以及患者預后均相關[10-11]。而基于ssGSEA免疫分型來分析免疫相關差異表達基因在肺腺癌預后方面價值的研究尚缺乏。
本研究基于TCGA數據集中肺腺癌患者樣本基因表達數據的ssGSEA免疫評分及Cox風險回歸分析,獲得肺腺癌預后相關的免疫差異表達基因。然后進行Lasso回歸及多元逐步Cox風險回歸分析降維,構建肺腺癌預后預測模型獲得Riskscore,并用GEO數據集進行驗證。利用Riskscore預測患者5年總生存期的AUC值均>0.7。將Riskscore與臨床特征進行獨立預后因子分析,并建立列線圖,列線圖1、3、5年的AUC也均>0.7。上述結果表明Riskscore及列線圖在預測肺腺癌總生存期上都具有良好的效能。
本研究結果顯示肺腺癌高免疫評分組的富集通路主要在“cytokine-cytokine receptor interaction”“natural killer cell mediated cytotoxicity”和“cell adhesion molecules”。而國內外也已有文獻報道這些通路在非小細胞肺癌中的發生、發展中起重要作用[12-14]。例如,Zheng等[12]研究結果表明在高免疫浸潤組的肺腺癌腫瘤樣本也主要富集在“cytokine-cytokine receptor interaction”。Zhang等[13]研究也發現EPHA5突變會破壞自然殺傷細胞介導的細胞毒性對非小細胞肺癌的作用,促進癌細胞遷移。Li等[14]研究表明高免疫評分組和低免疫評分組的差異基因富集通路也包括“cytokine-cytokine receptor interaction”和“cell adhesion molecules”。上述研究結果證實了這些通路可以作為肺腺癌潛在的免疫治療新靶點,進而提高肺腺癌患者預后水平。
本研究中,在對預后相關的免疫差異表達基因進行Lasso回歸及多元逐步Cox風險回歸分析降維后,獲得了最優的8個基因集合,其中權重最高的3個基因分別是TAP2、ARG2和CXCR6。這些基因在非小細胞肺癌以及肺腺癌進展方面的作用也已經有一些研究報道[15-17]。Liu等[15]研究結果表明TAP2的基因多態性與非小細胞肺癌的進展存在潛在的關系。Giatromanolaki等[16]研究結果發現ARG2主要在癌相關成纖維細胞中表達。說明這些基因具有預測肺腺癌患者預后的能力。但是,也有研究結果發現癌細胞中表達的CXCR16并沒有表現出對非小細胞肺癌預后的影響[17]。這可能是該研究納入的是非小細胞肺癌患者,與本研究中納入的肺腺癌患者不同所致,這需要進一步深入的研究來驗證。
通過類似的方法,Wang等[18]研究也是基于ssGSEA算法分析出的免疫相關差異表達的長鏈非編碼RNA(lncRNA)來構建肺腺癌預后的預測模型,其訓練集預測5年總生存期的AUC達到0.63,驗證集預測5年總生存期的AUC達到0.667。本研究則基于ssGSEA分析構建肺腺癌患者風險預測模型并獲得Riskscore,其在訓練集、內部驗證集及外部驗證集5年總生存期的AUC分別為0.703、0.713、0.750。同時將該Riskscore與臨床特征進行獨立預后分析并構建聯合模型列線圖,列線圖5年總生存期的AUC達到0.746。Riskscore與列線圖的AUC均較高且均>0.7,Riskscore的HR值也較臨床分期高,均進一步表明了Riskscore在預測肺腺癌預后方面的能力較好,同時該模型也具有較好的泛化性。通過校正曲線分析也進一步顯示了預測模型的結果與實際總生存期較為一致。
本研究存在一定的局限性。首先,研究納入的患者例數有限,僅用了來自于TCGA數據庫及GEO數據集,未來還需要多中心的數據來進行驗證。其次,本研究并未對篩選的風險基因進行相關機制研究,未來需開展基礎實驗進行進一步分析。
綜上所述,本研究基于ssGSEA分析獲得的免疫相關差異表達基因構建了肺腺癌風險預測模型,并聯合臨床特征繪制了列線圖,以期能夠輔助臨床醫師對肺腺癌患者總生存期進行判斷。
猜你喜歡
童話王國·奇妙邏輯推理(2024年5期)2024-06-19 16:03:38
音樂探索(2022年2期)2022-05-30 21:01:37
民用飛機設計與研究(2020年4期)2021-01-21 09:15:02
中學生數理化·七年級數學人教版(2020年10期)2020-11-26 08:24:50
數學物理學報(2020年2期)2020-06-02 11:29:24
小天使·一年級語數英綜合(2019年8期)2019-08-27 02:23:00
電子制作(2018年18期)2018-11-14 01:48:24
小學科學(學生版)(2018年7期)2018-08-13 09:33:04
山東工業技術(2016年15期)2016-12-01 05:31:22
光學精密工程(2016年6期)2016-11-07 09:07:19
