姜黃素自微乳結腸炎癥靶向微丸的處方研究
2022-09-15 07:19:20鞠佳芮戴俊東朱笑顏郭鵬川吳師月
長春中醫藥大學學報 2022年9期
關鍵詞:殼聚糖
鞠佳芮,戴俊東,朱笑顏,郭鵬川,吳師月
(北京中醫藥大學中藥學院,北京 102488)
潰瘍性結腸炎(UC)是一種慢性非特異性炎癥性腸病[1],病變部位主要集中于結腸黏膜或黏膜下層,呈彌漫性分布,常累及乙狀結腸和直腸[2]。由于病因不明,目前臨床上多以對癥抗炎治療為主。為改善其治療效果,在結腸靶向的基礎上,將藥物特異性靶向于結腸炎癥部位現已成為潰瘍性結腸炎給藥系統研究的重要方向。
UC炎癥部位轉鐵蛋白[3]、殺菌/通透性增強蛋白[4]、嗜酸細胞陽離子蛋白等陽離子蛋白表達異常增高[5],使得結腸炎癥部位帶正電荷。課題組前期制備的姜黃素荷負電自微乳,能夠顯著改善姜黃素的水溶性和在腸道部位的穩定性,并通過靜電吸附作用靶向于結腸炎癥部位[6]。為解決姜黃素荷負電自微乳釋藥迅速,直接口服給藥難以到達結腸部位的問題,本研究選擇殼聚糖與海藻酸鈉為丸芯材料,EudragitS 100包衣制備pH-酶依賴型結腸靶向微丸Cur-ITP,以實現姜黃素荷負電自微乳在結腸部位的靶向遞送和平穩釋放,對炎癥部位進行靶向治療。
1 儀器與試藥
BSA223S-CW電子分析天平(德國賽多利斯科學儀器有限公司);pH計[奧豪斯儀器(上海)有限公司];756PC型紫外可見分光光度計(上海舜宇恒平科學儀器有限公司);濕法制粒擠出滾圓一體機 CML(英國Caleva公司);SZX7體式顯微鏡(日本奧林巴斯株式會社);RC806D智能溶出儀(天津天大天發公司)。
姜黃素(純度95%,上海源葉生物科技有限公司,批號R26J7S18508);丁二酸二辛酯磺酸鈉(純度96%,上海麥克林生化科技有限公司,批號C10050237);丙二醇單辛酸酯(Capryol90,法國嘉法獅公司,批號144603);二乙二醇單乙基醚(Transcutol HP,法國嘉法獅公司,批號177728);聚氧乙烯氫化蓖麻油(CremophorRH40,上海源葉生物科技有限公司,批號Y17O11S127656);殼聚糖(脫乙酰度85%,分子量30~40萬,上海源葉生物科技有限公司,批號A14A11X121309);海藻酸鈉(化學純,粘度10 g·L-1≥0.02 Pa·S-1,西隴化工股份有限公司,批號120503);Eudragit S100(上海昌為醫藥輔料技術有限公司,批號B170105202);胃蛋白酶(30 000 U·g-1,上海源葉生物科技有限公司;批號:D20GS171943);胰蛋白酶(250 U·mg-1,上海源葉生物科技有限公司;批號:D29GS172831);β-甘露聚糖酶(50 000 U·g-1,上海麥克林生化科技有限公司,批號:C12318569)。
2 方法
2.1 微丸含量測定方法的建立
為便于微丸處方與釋放度研究大量樣品的快速檢測,在課題組前期HPLC含量測定方法[7]的基礎上,建立姜黃素微丸紫外—可見分光光度法含量測定方法。
2.2 姜黃素炎癥靶向自微乳的制備[6]
取 Cremophor RH40 6.0 g,Transcutol HP 3.0 g,Capryol 90 1.0 g,丁二酸二辛酯磺酸鈉0.4 g,70℃水浴加熱攪拌并使之完全溶解,然后加入姜黃素0.4 g,100 r·min-1攪拌15 min,得姜黃素炎癥靶向自微乳,加水稀釋后平均粒徑(19.75±0.43)nm,Zeta電位為(-40.47±1.68)mV。
2.3 微丸丸芯處方研究
以處方中殼聚糖與海藻酸鈉的比例(A)、姜黃素自微乳用量(B)、潤濕劑用量(C)為影響因素;合格微丸收率(Y1)、載藥量(Y2)、圓整度(Y3)和釋放度(Y4)為指標,進行Box-Behnken設計(見表2)。按處方量(總質量均為12 g)稱取殼聚糖與海藻酸鈉,加入姜黃素自微乳混勻,再加入水為潤濕劑制軟材,擠出—滾圓法制備微丸,擠出速度100 rpm;滾圓速度2 000 rpm,60℃烘箱干燥即得。
2.4 微丸質量評價
2.4.1 合格微丸收率(Y1) 利用篩分法計算能通過一號篩不能通過二號篩的微丸收率。
2.4.2 載藥量(Y2) 取適量微丸研細,精密稱取0.015 g,加pH 1.0鹽酸溶液10 mL,超聲提取70 min,冷卻,取1 mL置于5 mL容量瓶中,乙醇定容,搖勻,8 000 rpm離心,取上清液測定姜黃素含量[8],計算載藥量。
2.4.3 圓整度(Y3) 隨機選取9粒微丸,體式顯微鏡測定其長寬比,計算其平均值與1的差值,即Y3=│X-1│,Y3值越小,微丸的圓整度越高。
2.4.4 釋放度(Y4) 采用37℃恒溫水浴振蕩法測定微丸的釋放度[9]。稱取姜黃素微丸(含姜黃素0.5 mg),加入pH7.4磷酸緩沖液15 mL,60 rpm恒溫振蕩[10],分別于15 min、30 min、1 h、2 h、4 h取1 mL上清液置于5 mL容量瓶中,依法測定,計算釋放度。為減緩自微乳中表面活性劑和助表面活性劑對結腸黏膜炎癥部位的刺激,實現微丸的平穩釋放,將30 min、1 h、2 h的釋放度分別與期望釋放值25%、50%、80%做差值,用偏差之和評價其釋放效果,即Y4(%)=│Q30min-25%│+│Q1h-50%│+(80%-Q2h)。
2.5 結腸靶向微丸的制備
按表1處方配制包衣液,對最佳工藝制得的丸芯進行包衣,至包衣增重達到12%~13%后80 ℃熱風干燥30 min,使衣膜塑化,形成連續致密的包衣層。
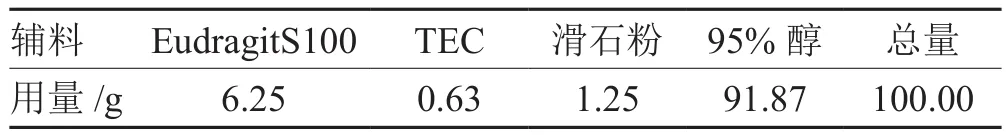
表1 包衣液處方
2.6 結腸靶向微丸釋放度研究
稱取姜黃素微丸(含姜黃素5 mg),采用小杯籃法,轉速50 rpm,釋放液體積150 mL,模擬體內胃腸道條件,依次考察包衣微丸在不含酶的pH 1.0、pH 4.5、pH 5.5、pH 6.8、pH 7.4釋放液和含酶的pH 1.0人工胃液(含胃蛋白酶);pH 4.5、pH 5.5、pH 6.8人工腸液(含胰酶);pH 7.4人工結腸液(含β-甘露聚糖酶)中的累積釋放度,分別于2 h、4 h、6 h、7 h、8 h、9 h、10 h、12 h取樣測定,評價包衣微丸的結腸靶向效果。
3 結果
3.1 微丸含量測定方法的建立
微丸經pH1.0鹽酸溶液超聲提取,可以使殼聚糖充分溶解,釋放與其通過靜電吸附的姜黃素荷負電自微乳,再經乙醇溶解破乳后于423 nm測定姜黃素含量。經方法學研究,溶劑與微丸輔料對姜黃素的含量測定無干擾,姜黃素濃度在1.9 992~4.9 980 μg·mL-1范圍內與吸光度線性關系良好,溶液穩定性、精密度、重現性和回收率考察結果均符合要求,可用于姜黃素微丸的含量測定。
3.2 Box-Behnben中心組合方法試驗設計及結果
見表2。
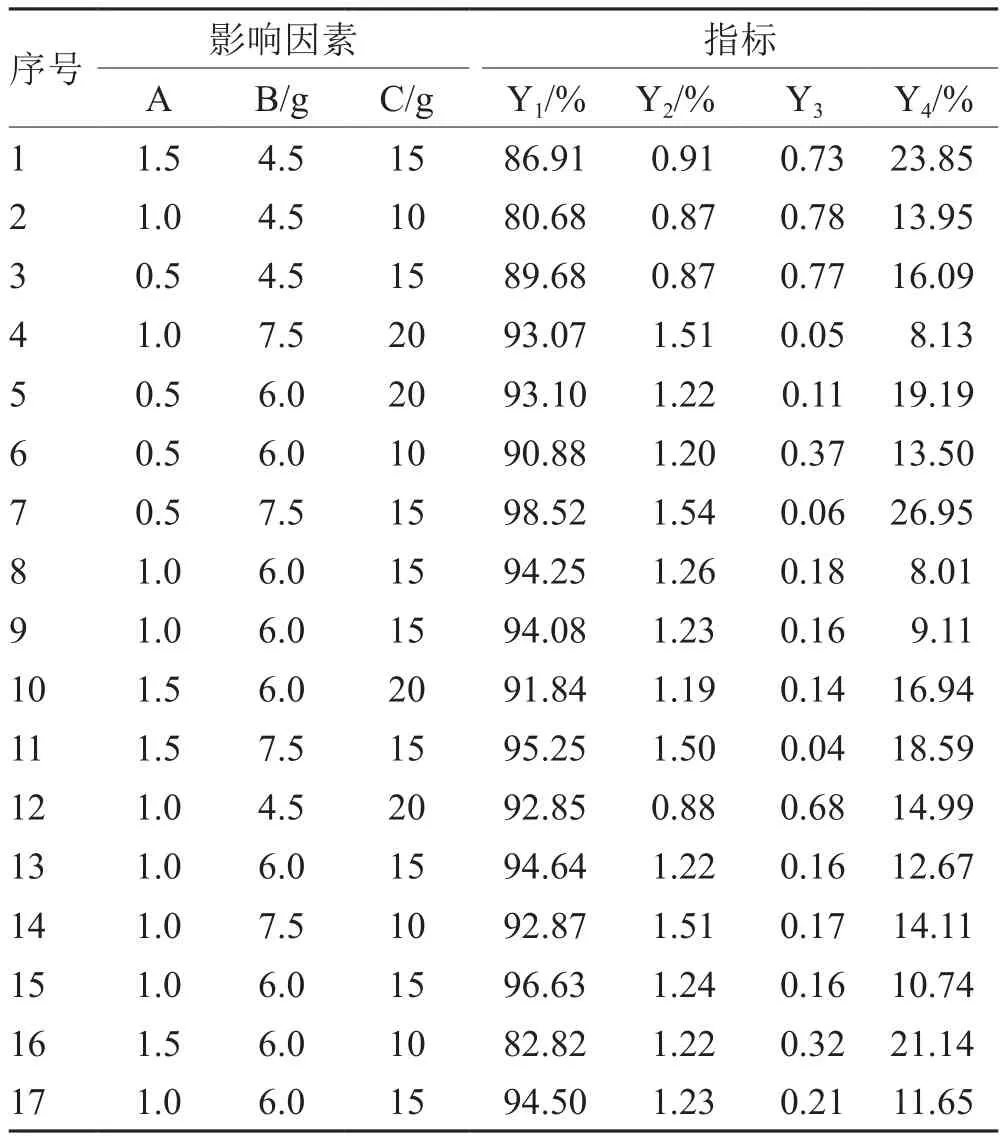
表2 Box-Behnben中心組合方法試驗設計及結果
3.3 模型組合
表2數據經多元回歸擬合,得到二項式擬合方程。F值均具有顯著性且失擬項不顯著,相關系數r2>0.9,說明擬合效果準確可靠。見表3。
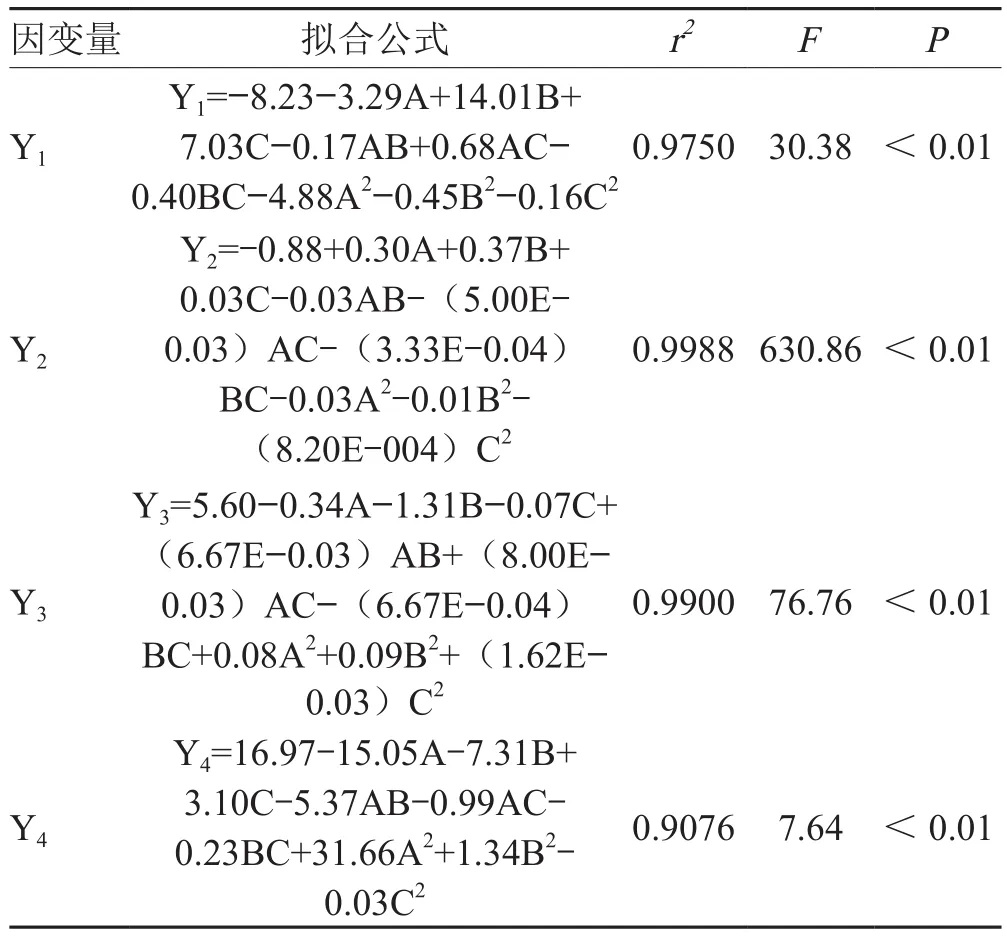
表3 模型擬合結果
3.4 模型分析[11]
根據擬合方程,繪制Cur-ITP處方優化指標隨因素變化的效應面圖。結果表明,A、B、C三個因素均對合格微丸收率Y1有顯著性影響(P< 0.01)。在B值一定的情況下,A值越小,即海藻酸鈉比例越高,潤濕劑水加入后誘導出的黏性越大,合格微丸的收率越高;但當B、C值均較高時,制得的軟材黏度過高,擠出后滾圓時易滾成大丸,導致符合粒徑要求的微丸收率降低。
僅B因素對Y2有顯著性影響(P<0.01),即處方中姜黃素自微乳用量越大,微丸的載藥量越高。但姜黃素自微乳的用量會受到處方中潤濕劑水的用量和固體粉末殼聚糖與海藻酸鈉用量的制約。總液體量過大,固體粉末難以吸收;姜黃素自微乳用量大,水量少則軟材粘性不足,可塑性差,難以成丸。姜黃素自微乳用量小,水量大則軟材粘性過大,亦難以成丸。因此,需要在保證軟材與成丸質量的前提下盡可能提高處方中姜黃素自微乳的用量。
B、C兩因素對Y3有顯著性影響(P<0.01),即姜黃素自微乳與潤濕劑水的用量對微丸的圓整度影響顯著。加入的總液體量越多,軟材可塑性越好,Y3值越小,圓整度越高。如前所述,總液體量會受到處方中固體粉末殼聚糖與海藻酸鈉用量的制約,姜黃素自微乳與潤濕劑水的比例對軟材的可塑性與微丸的成型和圓整度亦有重要影響。
A2對Y4有顯著影響(P<0.01),表明因素A即處方中殼聚糖與海藻酸鈉的比例對微丸的釋放度影響顯著。當A值較高時,微丸中海藻酸鈉用量較少,導致微丸溶散速度較快,突釋效應明顯,30 min時姜黃素的釋放度約30%,且由于微丸中大量殼聚糖的靜電吸附作用使2 h時姜黃素的釋放度低于70%,緩釋作用明顯;當A值較低時,海藻酸鈉遇水形成的凝膠控制微丸30 min時姜黃素的釋放度低于20%。30 min~1 h時海藻酸鈉逐漸溶于釋放介質,而微丸中殼聚糖的用量較少,靜電吸附作用較弱,使1 h時姜黃素的釋放度高于50%,均不符合姜黃素平穩釋放的預期。故需要根據預期釋放特征,合理確定微丸處方中殼聚糖與海藻酸鈉的比例。因素與自變量A-C的曲面圖,見圖1。
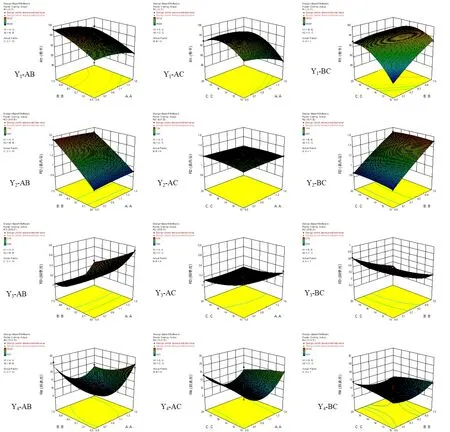
圖1 因素與自變量A-C的曲面圖
3.5 驗證試驗
選取Y1、Y2最大,Y3、Y4最小為最優條件,按處方中殼聚糖:海藻酸鈉1.07、自微乳7.50 g、水16.86 g制備3批Cur-ITP,依法測定各項指標,所得實測值與預測值基本一致(見圖2、表4、圖3),說明模型預測性良好,處方工藝穩定。
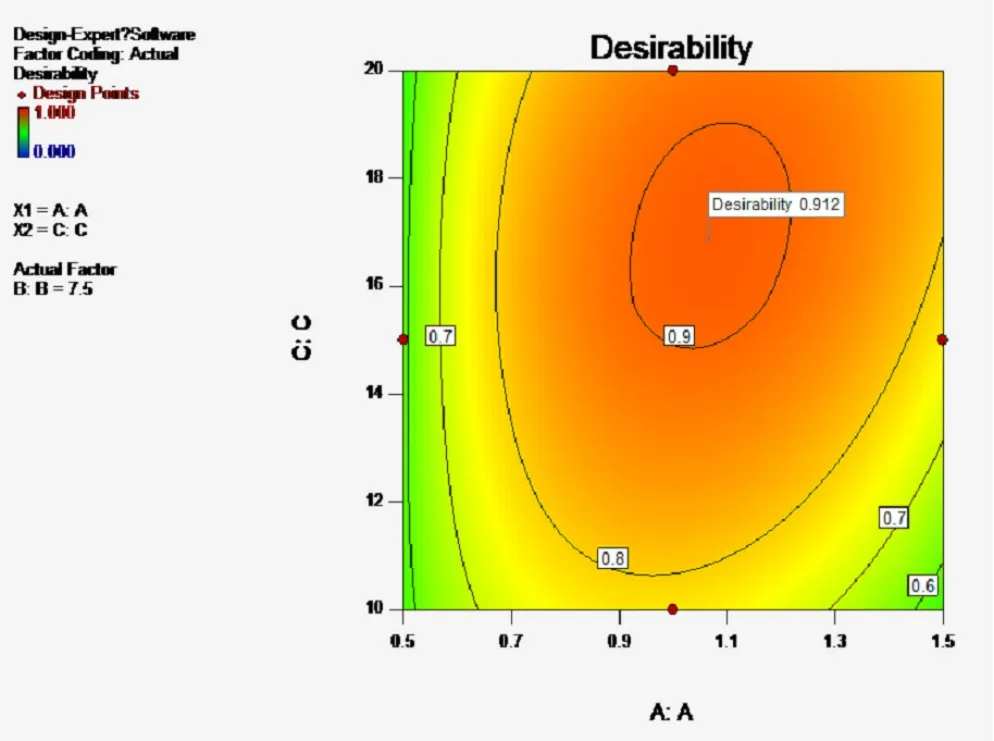
圖2 Cur-ITP制備工藝預測值
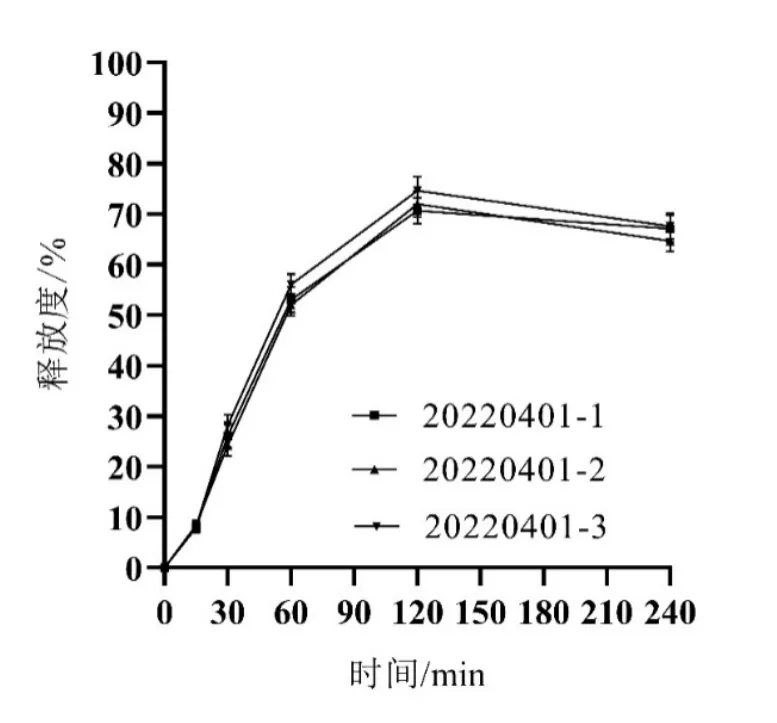
圖3 最優處方微丸釋放曲線
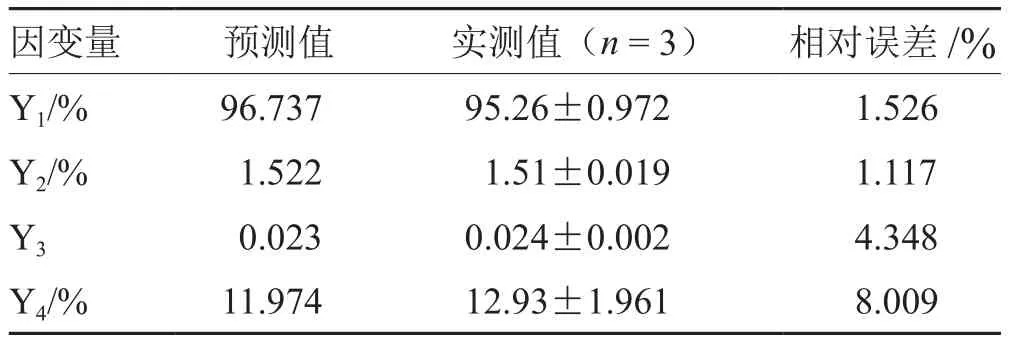
表4 最優處方驗證實驗
3.6 結腸靶向微丸釋放度
由圖4可見,包衣微丸在pH 4.5以下的模擬釋放介質中基本不釋放,pH 6.8以下,包衣膜基本保持完好,含酶與不含酶的釋放介質對包衣微丸的釋放度影響不大,姜黃素在pH 6.8的模擬釋放介質2 h后釋放度低于25%,說明包衣微丸可使藥物到達小腸末端再釋放,結腸靶向性良好。pH 7.4條件下,包衣膜溶解,姜黃素在含酶的人工結腸液中釋放度增加,這主要與加入的β-甘露聚糖酶對殼聚糖的分解作用有關。
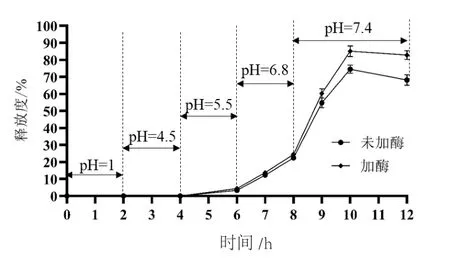
圖4 結腸靶向微丸累積釋放度曲線
4 討論
殼聚糖與海藻酸鈉均為天然多糖,在體內具有良好的生物相容性與生物可降解性[12-13],兩者常采用乳化交聯[14]和離子交聯[15]等方法制備結腸靶向微球[16]。本實驗前期嘗試將姜黃素荷負電自微乳與海藻酸鈉混合后噴霧干燥[17],制得的微粉再與殼聚糖溶液進行離子交聯制備微球。但由于自微乳中含有較多表面活性劑和助表面活性劑,在交聯時姜黃素向水中快速擴散,導致姜黃素微球包封率及載藥量均較低。并且微球的突釋效應明顯,10 min即可釋放55%~60%。為解決上述問題,本實驗采用殼聚糖與海藻酸鈉固體粉末吸附姜黃素荷負電自微乳,再以水為潤濕劑制軟材,擠出—滾圓法制備微丸,取得了較好的效果,具有工藝簡便,載藥量高,釋放平穩的優點。
本研究結果表明,Cur-ITP的釋放效果和處方中殼聚糖與海藻酸鈉的比例密切相關。海藻酸鈉比例越高,微丸在溶散前釋放度越低,但溶散后釋放速率較快,釋放度最大值較高;殼聚糖比例越高則釋放度最大值越低,釋放過程中速率越平緩。究其原因,主要與海藻酸鈉、殼聚糖的性質和兩者與姜黃素荷負電自微乳間的靜電相互作用有關。海藻酸鈉為羧酸鈉鹽,在pH7.4的磷酸緩沖液中可溶脹形成凝膠,繼而逐漸溶蝕,電離后的羧酸根在溶液中帶有負電荷[18],與荷負電自微乳間存在電荷排斥作用。因此,海藻酸鈉主要通過溶脹后形成凝膠的粘度和溶蝕速度控制微丸的溶散速度和藥物的擴散,對微丸的突釋效應影響顯著。而殼聚糖為氨基多糖,分子量較海藻酸鈉更大,在pH7.4的磷酸緩沖液中溶脹很小,產生的粘性較低,故對微丸的溶散速度影響較小。但其表面的氨基在pH7.4的磷酸緩沖液中帶有正電荷[19],與荷負電自微乳間產生電荷吸附作用,可以通過大分子的空間網格結構,以及靜電引力控制荷負電自微乳的擴散,從而對微丸溶散后姜黃素的釋放起主要控制作用。通過在包衣微丸的累積釋放度實驗中模擬結腸菌群的作用[20],即在pH7.4的磷酸緩沖液中加入β-甘露聚糖酶[21],使殼聚糖酶解,可顯著增加微丸溶散后姜黃素的釋放度。
此實驗結果驗證了上述推論的合理性。
猜你喜歡
河北科技師范學院學報(2022年2期)2022-08-26 08:55:40
河北科技師范學院學報(2021年1期)2021-05-10 03:34:20
中成藥(2017年12期)2018-01-19 02:06:57
電源技術(2017年1期)2017-03-20 13:37:59
廣西科技大學學報(2016年1期)2016-06-22 13:10:38
天然產物研究與開發(2016年1期)2016-06-05 10:29:25
食品界(2016年4期)2016-02-27 07:36:46
中國果菜(2015年2期)2015-03-11 20:01:01
應用化工(2014年7期)2014-08-09 09:20:21
應用技術學報(2014年4期)2014-02-28 14:52:40
