不同表達矩陣對篩選差異長鏈非編碼RNA的影響
2022-09-19 02:40:28邱家俊顏景斌
上海交通大學學報(醫(yī)學版) 2022年7期
魏 豪,邱家俊,顏景斌
上海市兒童醫(yī)院,上海交通大學醫(yī)學院附屬兒童醫(yī)院醫(yī)學遺傳研究所,上海市胚胎與生殖工程重點實驗室,上海 200040
長 鏈 非 編 碼RNA (long non-coding RNA,lncRNA)是指一類核苷酸長度大于200nt 的非編碼RNA,與其他的非編碼RNA 曾一度被認為是“轉錄噪聲”[1]。隨著第二代測序技術的發(fā)展,人們發(fā)現(xiàn)在許多模式生物以及人類的基因組中,lncRNA 均有廣泛的表達。同時越來越多的功能研究認為lncRNA 可以參與調節(jié)多種重要的細胞活動,如基因表達、招募染色質修飾物、調節(jié)X染色體失活、基因組印記、蛋白質折疊和蛋白質活性等[2]。
以第二代測序技術為基礎的全轉錄組測序逐漸成為研究lncRNA 表達和功能的重要手段。通過對轉錄組測序數據的處理和分析,人們可以更加具體地在轉錄本異構體、核苷酸變異、轉錄后堿基修飾等方面進行研究。其中的一個主要環(huán)節(jié)就是對不同組別之間的測序數據進行差異表達分析,表達水平的準確估計對這個過程至關重要[3]。
在轉錄組測序中,計算一個轉錄本的表達水平往往要考慮到測序深度、基因長度等因素。起初人們選擇使用每百萬reads 中每1 000 堿基長度的reads 數(reads per kilobase per million mapped reads,RPKM)等標準化數值來表示相對表達水平,以減少非研究因素的影響,但仍有一些不足,例如在差異分析時部分基因表達水平過高就會產生大量的假陽性結果[4-6]。此外人們還開發(fā)出許多R 語言軟件包,例如DESeq2[7]、edgeR[8]、PoissonSeq[9]、CuffDiff[10]等,這些軟件往往采用自研的一套算法直接對原始表達數據進行標準化處理,同時分析差異基因表達水平。
在分析全轉錄組測序中l(wèi)ncRNA 的差異表達水平時,人們往往直接對全轉錄組表達矩陣進行差異分析,再從差異分析結果中將lncRNA 分選出來。但許多研究已經表明lncRNA 的表達水平普遍低于編碼蛋白質的mRNA[1],這就意味著lncRNA 和mRNA 的表達水平分布或許也不相同,以此為背景對全部RNA的表達數據進行標準化和差異分析時,就可能導致一部分沒有表達差異的lncRNA 被認為具有差異,產生假陽性結果。為了減少差異分析時各種背景因素的影響,人們選擇使用DESeq2 或edgeR 中的標準化模型進行分析。已有不少研究表示,DESeq2 和edgeR 差異表達分析的假陽性率(false positive rate,F(xiàn)PR)更低[11]。因此,選擇合適的分析方法對于篩選差異的lncRNA至關重要。
本研究對全轉錄組測序中僅具有l(wèi)ncRNA 的表達矩陣進行差異表達分析,并將之與總體RNA 表達矩陣的分析結果作比較,從而探究出在篩選差異lncRNA方面誤差更小的方法。
1 數據與方法
1.1 數據來源
從NCBI_GEO 數據庫(https://www.ncbi.nlm.nih.gov/geo)中下載2個不同來源的人類全轉錄組測序數據(GSE49712),共10個樣本。A組為5個人類通用參考RNA(universal human reference RNA,UHRR)樣本,每個樣本摻入了2%體積的外源RNA對照物聯(lián)盟(external RNA control consortium,ERCC)混合物1;B組為5個人腦參考RNA(human brain reference RNA,HBRR)樣本,每個樣本摻入了2%體積的ERCC混合物2。ERCC spike-in 對照是由92 種含polyA 序列的人工合成的外源寡核苷酸組成,它們在A組和B組之間的摻入比例有以下4種:1∶2、2∶3、1∶1和4∶1。
從賽默飛網站(https://www.thermofisher.cn)中下載ERCC spike-in 對照的fastq 文件和GTF 文件。從GENCODE 網站(https://www.gencodegenes.org)中下載人類GRCh37 完全注釋基因組以及相應的lncRNA注釋基因組。
1.2 獲取表達矩陣
在獲得原始SRA 格式的數據后,對其進行格式轉換、質控(去除低質量reads和接頭)、比對和計數等處理,具體步驟如下。
(1)使用sratoolkit(version 2.10.8)程序將原始數據轉換為fastq格式。
(2)使用trim_galore(version 0.6.6)程序過濾掉低質量reads;使用FastQC程序查看質控結果。
(3)使用hisat2(version 2.2.1)程序中的hisat2-build 命令構建含有spike-in 序列的參考基因組,然后使用hisat2命令對reads進行比對。
(4)使用samtools (version 1.7)程序對獲得的二進制sam文件進行排序、建立索引等處理。
(5)使用awk 命令從人類GRCh37 完全注釋基因組中提取出僅含有編碼蛋白基因的注釋基因組,再將spike-in的注釋信息分別加入各個注釋基因組中。
(6)使用featureCounts(version 2.0.1)程序對比對后的片段進行計數和注釋,分別獲取總體RNA 表達矩陣、mRNA表達矩陣和lncRNA表達矩陣。
1.3 表達水平差異分析
分別使用DESeq2 和edgeR 軟件包對A 組和B 組之間的差異表達水平進行分析,獲取以3 種表達矩陣為背景的差異spik-in 信息。使用DESeq2 分別對A 組和B組組內的差異表達水平進行分析,獲取總體RNA表達矩陣和lncRNA表達矩陣的差異lncRNA信息。
本研究要比較的2 種分析方法分別為使用總體RNA 表達矩陣篩選差異lncRNA 的方法(簡稱總體RNA 法)和使用lncRNA 表達矩陣篩選差異lncRNA的方法(簡稱lncRNA法)。
1.4 樣本層級聚類分析
分別過濾掉每個表達矩陣中沒有表達的基因,使用R 語言中的hclust 功能對各個表達矩陣中所有樣本進行層級聚類分析。
1.5 受試者操作特征曲線(ROC)分析
使用pROC 軟件包對3個表達矩陣的差異spike-in信息進行分析,獲取在不同P值下的FPR和真陽性率(true positive ratio,TPR),并計算其曲線下面積(area under the curve,AUC)值。其中在A 組和B 組之間摻入比例為1 的可認定為沒有差異,其余3 種比例則認定為有差異。
1.6 統(tǒng)計學方法
對不同方法獲取差異spike-in 信息效果的評價采用ROC 曲線,使用AUC 值預測特異性和準確性。部分所得數據使用Origin 2022 軟件進行統(tǒng)計學分析并作圖。P<0.05表示差異有統(tǒng)計學意義。
2 結果
2.1 表達矩陣的獲取
本研究對2個組別共10個樣本的測序數據進行處理之后,使用不同的注釋基因組對其計數,獲得了3個表達矩陣(總體RNA、mRNA、lncRNA)。經統(tǒng)計,A 組樣本和B 組樣本中分別有47 583 個和47 177 個已知基因得到了表達,其中A 組樣本包括19 061 個mRNA 基因和15 711 個lncRNA 基因,B 組樣本包括18 884個mRNA基因和15 652個lncRNA基因。A組樣本中mRNA 和lncRNA 總體表達水平的平均占比分別為95.58%和3.12%,B 組樣本中mRNA 和lncRNA 總體表達水平的平均占比分別為93.87%和3.71%。我們對各個表達矩陣中的樣本進行了層級聚類分析,結果表明所有表達矩陣均能很好地分辨樣本差異(圖1)。
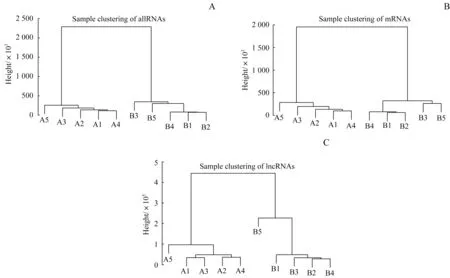
圖1 A組和B組樣本表達矩陣的層級聚類圖Fig 1 Hierarchical clustering of the expression profiles from sample A and B
2.2 Spike-in表達水平差異分析
使用DESeq2軟件包對3個表達矩陣進行差異表達分析,從中提取出spike-in在A組和B組之間的差異表達信息。為了確認在廣泛采用的差異基因篩選標準(P<0.05) 下,總體RNA 法和lncRNA 法篩選差異lncRNA的效果,我們統(tǒng)計了spike-in在總體RNA表達矩陣和lncRNA 表達矩陣中假陽性和假陰性的數目(表1),并計算了FPR 和假陰性率(false negative rate,F(xiàn)NR)(圖2A)。結果發(fā)現(xiàn),使用總體RNA法分析的spike-in FPR 為0.52(12/23),而使用lncRNA 法分析的spike-in FPR 為0.30(7/23),顯然后者篩選差異spike-in的特異性更好。我們對總體RNA 表達矩陣中多出的5個假陽性spike-in進行了進一步分析,除了其中一個spike-in 表達水平極低以外,其余4 個spikein的表達數據在經過標準化處理之后,總體RNA表達矩陣比lncRNA表達矩陣的組間差異更大(圖2B)。
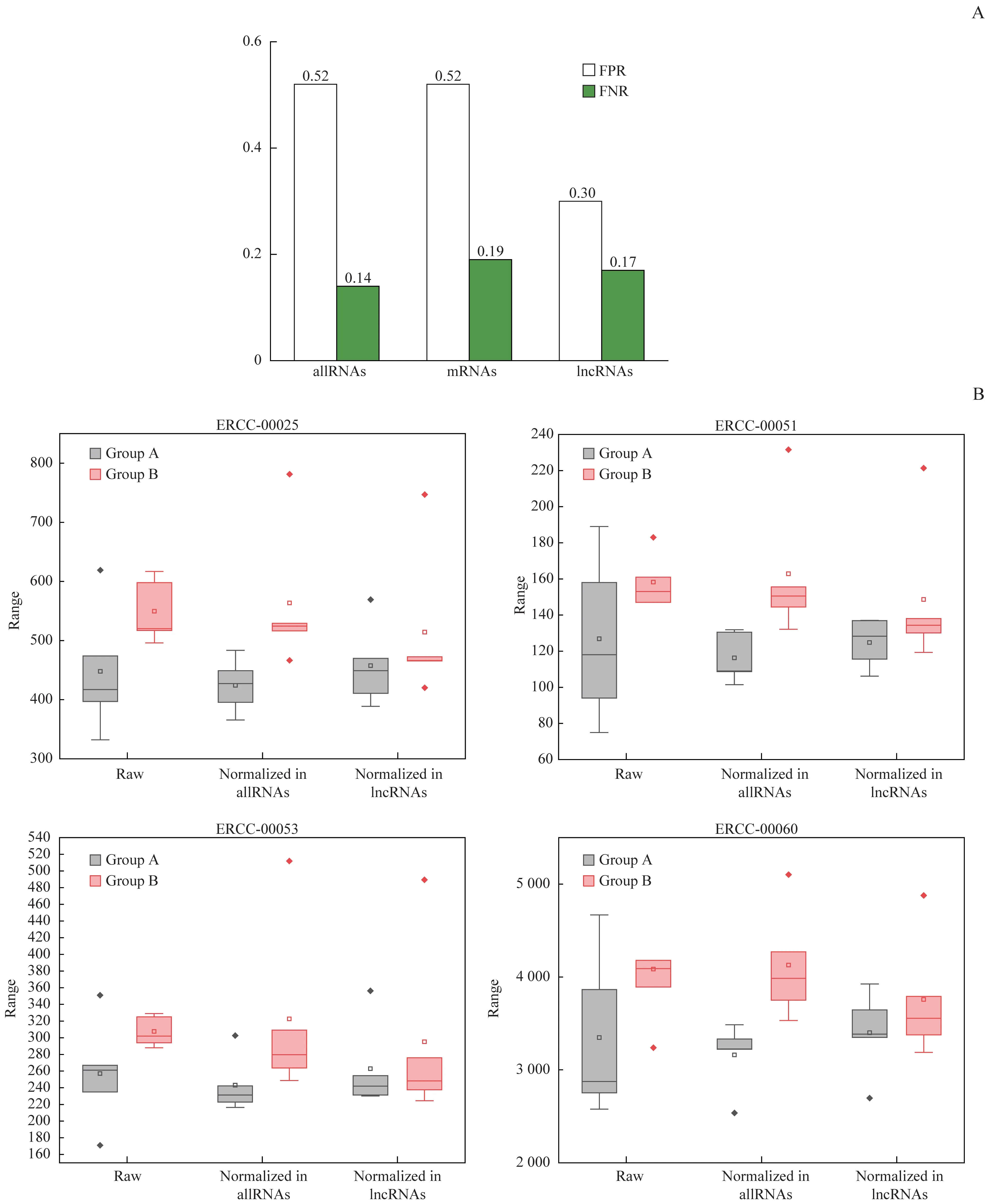
圖2 Spike-in RNAs差異表達分析的結果Fig 2 Differential expressions of spike-in RNAs
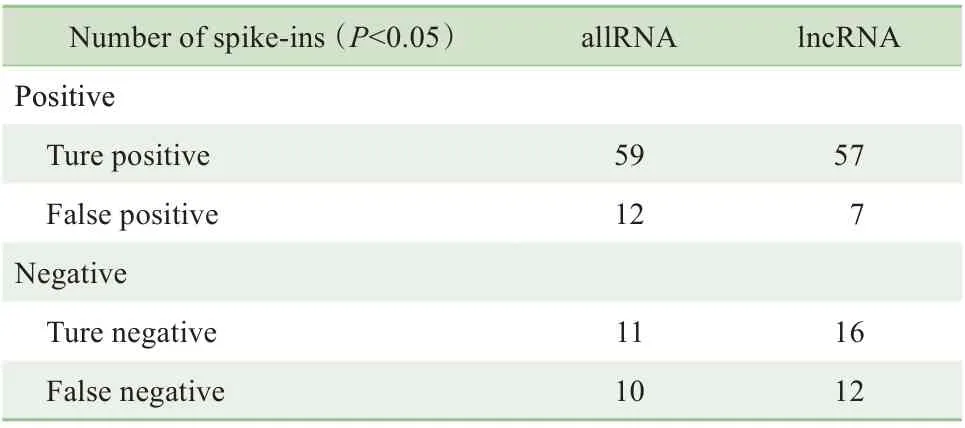
表1 2個表達矩陣預測的差異表達spike-in RNAs數目Tab 1 Number of differential expression spike-in RNAs predicted by two different expression profiles
2.3 ROC分析
為了進一步觀察其他P值條件下的差異基因篩選效果,本研究采用spike-in 在不同表達矩陣背景下的P值作ROC 曲線。同時為了排除算法因素的影響,我們分別使用DESeq2 和edgeR 進行ROC 分析(圖3),其中橫坐標和縱坐標分別用特異性和準確性表示,它們在數值上分別與1-FPR 和TPR 相等。通過每條ROC 曲線下的面積大小,即AUC 值來量化不同分析方法篩選差異spike-in 的效果。在DESeq2 的分析結果中,Spike-in 在3 個表達矩陣中的AUC 值大小關系為AUC(lncRNA)=0.852>AUC(allRNA)=0.768>AUC(mRNA)=0.750,而在edgeR 的分析結果中為AUC(lncRNA)=0.878>AUC(mRNA)=0.798>AUC(allRNA)=0.787。顯然,spike-in 以lncRNA 表達矩陣為背景的AUC值顯著高于其他2個表達矩陣。
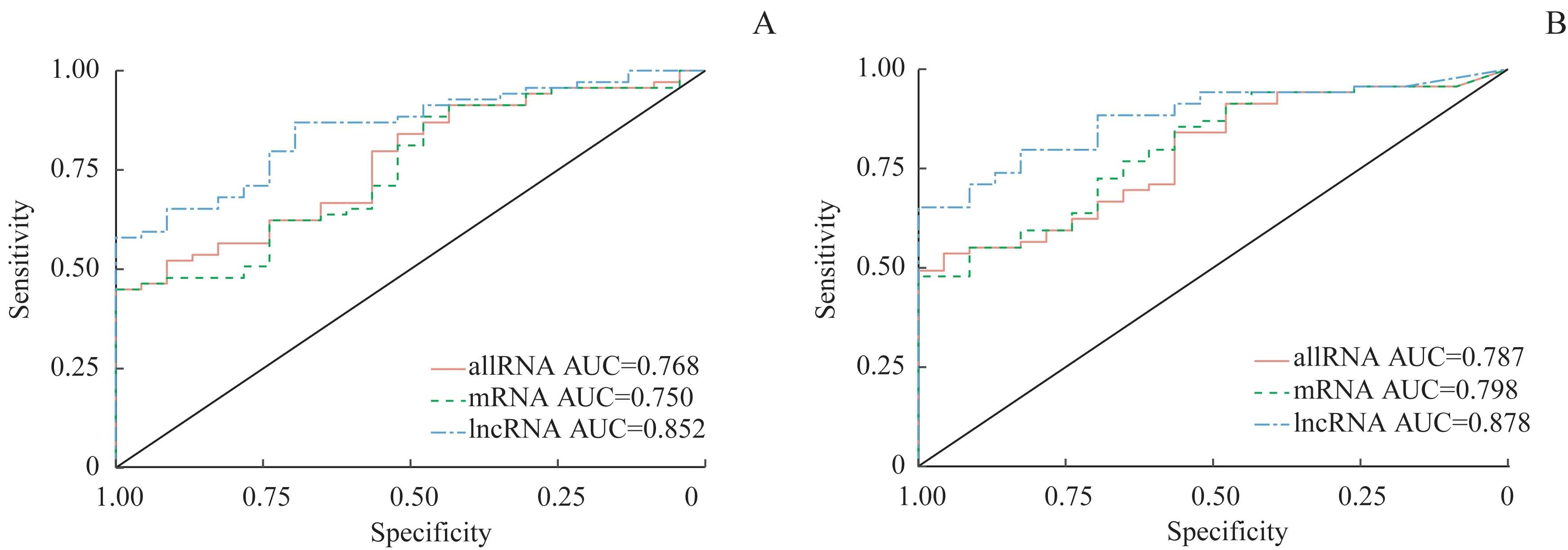
圖3 使用DESeq2和edgeR對spike-in RNAs的ROC分析結果Fig 3 ROC curve of ERCC spike-in RNAs analyzed by DESeq2 and edgeR
2.4 差異表達lncRNA的篩選
由于組內各個樣本的測序數據來源完全相同,理論上可以認為組內樣本之間基因表達水平沒有差異,因此可以用來評估差異分析方法的FPR大小。本研究使用DESeq2 對總體RNA 表達矩陣和lncRNA 表達矩陣中的A 組和B 組分別進行組內差異lncRNA 分析,其分組為A1 和A2、A3 和A4、B1 和B2、B3 和B4。由于組內樣本絕大多數基因無表達差異,P值無限接近于1,因此我們在對lncRNA 的P值作密度分布圖時,把范圍縮小在0~0.2之間。當把篩選差異lncRNA的標準定為P<0.05 時,在A 組中l(wèi)ncRNA 表達矩陣和總體RNA 表達矩陣的差異lncRNA 數目分別為9 個(占比2.57%)和7 個(占比2.45%),見圖4A;B 組中分別15 個(占比3.46%)和17 個(占比3.65%),見圖4B。使用不同表達矩陣篩選差異lncRNA 的FPR并沒有太大差別,可能與本研究組內樣本之間差異基因數目過少有關。

圖4 組內差異表達lncRNAs的P值密度曲線Fig 4 Density curve of P-values of differential expressed lncRNAs within groups
2.5 mRNA和lncRNA的表達水平分布
為了進一步探究使用總體RNA 法差異分析的FPR 更高的原因,我們對mRNA 和lncRNA 的表達水平分布進行了分析。鑒于不同基因之間的原始表達水平(raw counts)差距過大,我們對其作對數化處理后進行展示。可以看到在全部樣本中mRNA 和lncRNA 各自表達水平的分布基本一致(圖5A),而mRNA和lncRNA之間則明顯不同(圖5B)。

圖5 mRNA和lncRNA表達水平的分布圖Fig 5 Distribution of mRNA and lncRNA expression levels
3 討論
近些年來,第二代測序技術以其不斷提高的技術水平和持續(xù)降低的成本快速發(fā)展,以之為基礎的轉錄組測序也成為大部分研究人員進行基因表達研究的主要選擇之一[12]。在鑒定不同環(huán)境下或不同組織之間樣本的差異表達基因方面,相比于之前發(fā)展起來的基因芯片等技術,轉錄組測序有著明顯的優(yōu)勢,例如可以對整個基因組實現(xiàn)更高的覆蓋率以及可以容易地檢測到低表達基因等[13]。但同時轉錄組測序數據的分析難度也大大提高了,在進行差異表達分析時,除了要考慮基因長度和測序深度外,其他基因的表達水平也是一個重要因素,部分過高表達的基因會影響低表達基因差異程度的評估[5]。而在全轉錄組測序中大部分lncRNA 表達水平往往明顯低于mRNA,因此在篩選差異lncRNA 時,難免會受到其他高表達mRNA的影響。因此,選擇合適的篩選方法對于lncRNA 的研究具有重要意義。
自轉錄組測序誕生以來,圍繞差異表達基因分析方法的研究不斷涌現(xiàn)[14],這些研究往往是對差異分析之前的一個關鍵步驟——數據標準化方法的改進和創(chuàng)新。盡管最初有人認為轉錄組測序并不需要復雜的標準化過程[15],但實際分析時總是需要在樣本之間進行對比,而原始reads數目受到的非研究因素影響過多,顯然不能直接用來比較。最初得到廣泛應用的轉錄組標準化數值為MORTAZAVI 等[16]開發(fā)的RPKM。RPKM 值對基因長度和測序深度作了標準化處理,但缺點也很明顯,每個基因RPKM值大小的計算很大程度上受到其他基因表達水平的影響,這可能產生實際并不存在的表達差異。隨后出現(xiàn)的其他相對表達量均有類似的問題,但這并不影響它們替代原始表達數據被廣泛使用。后來人們開發(fā)的標準化算法往往與差異表達算法一起整合在軟件包中,它們基于大部分基因的表達水平是不具有差異的這一假設,通過各自的方式計算出歸一化因子來對數據進行標準化處理。代表性的有DESeq2所使用的RLE(relative log expression)算法[7]和edgeR所使用的TMM(trimmed mean of Mvalues)算法[8]。理論上這2種標準化方式能夠較好地減少高表達基因或高變基因的影響。因此,本研究使用DESeq2和edgeR軟件包執(zhí)行差異分析過程,在另一方面也可以來檢驗這2種標準化方式的實際效果。
本研究從NCBI_GEO數據庫中下載了2組來源于測序質量控制聯(lián)盟(Sequencing Quality Control Consortium,SEQC)的人類參考RNA測序數據[17-18]。由于這2 組數據中包含不同摻入比例的已知濃度的ERCC spike-in對照,以及近1 000個已在qPCR實驗中驗證表達的基因,常常也被用于轉錄組測序分析方法的研究。在獲得了包含spike-in表達信息的3個表達矩陣后,過濾掉在所有樣本中均不表達的基因,我們發(fā)現(xiàn)表達mRNA 的基因比表達lncRNA 的基因數目僅多出3 000~4 000 個,而mRNA 在全基因組水平的表達占比上卻遠遠大于lncRNA,說明絕大多數lncRNA的表達水平普遍低于mRNA。對分開的表達矩陣層級聚類分析結果表明,僅有l(wèi)ncRNA 的表達矩陣也能對不同樣本作出良好區(qū)分,這與已知許多證明lncRNA 更具有組織特異性的研究基本一致[19-20]。
在差異分析后基于每個spike-in 的P值作出的ROC 曲線來看,不管使用哪個軟件包執(zhí)行差異分析,在lncRNA 表達矩陣中的AUC 值都明顯大于另外2 個表達矩陣,而由于mRNA 在所有RNA 中表達占比極高,它們的AUC 值大小不相上下。說明在篩選差異lncRNA時,選擇使用lncRNA法進行差異分析更接近實際的差異表達情況。我們通過對使用總體RNA 法多出的假陽性spike-in 進一步分析發(fā)現(xiàn),它們的表達數據在總體RNA 表達矩陣背景下的標準化數值組間差異更大,這意味著即使使用DESeq2 或edgeR 等這類在標準化處理時考慮到高表達基因和高變基因的軟件包時,也難以完全避免它們的影響。本研究對組內總體RNA 表達矩陣和lncRNA 表達矩陣的差異lncRNA 分析結果表明,在P<0.05 的篩選條件下,使用2 種方法分析的FPR 非常接近,這與前面篩選差異spike-in 的研究結果不太一致,但仍能說明直接使用lncRNA表達矩陣篩選差異基因的方法是可行的。
值得注意的是,本研究中篩選差異基因的標準均設為P<0.05,這是因為在很多研究中該標準被看作是差異具有顯著性的分水嶺[21]。該標準最早由FISHER R在20世紀20年代中期提出,用來描述農業(yè)田間試驗的顯著性[22]。后來該標準應用于醫(yī)學領域之后,更高的犯錯成本使得人們不得不重視它所帶來的諸多問題。首先,部分研究者不惜采取P值操縱(P-hacking)的方式來使結果差異更大[23],這引起了更多研究人員的不滿。其次,P<0.05 的統(tǒng)計結果意味著仍有約5%的概率得到假陽性結果,而高通量測序數據中動輒上萬的基因就會產生相當數目的假陽性結果,大量的差異基因也使得FPR比預估的更高[24]。本研究也間接證實了這一點,該標準下篩選的差異spike-in FPR 都比較高。為了降低FPR,許多轉錄組差異分析軟件額外采用了padj(adjustP-value)來表示 顯 著 水 平[25]。padj 是 經 過FDR (false discovery rate)矯正后的P值。本研究在對組內樣本執(zhí)行差異分析時也發(fā)現(xiàn),所有基因的padj 均無限接近于1,即沒有假陽性。盡管如此,在實際研究過程中P<0.05仍是表明研究結果具有顯著意義的必要條件,但也應注意不能濫用或過于偏信該標準。
我們通過以結果為導向的研究反推出了使用lncRNA表達矩陣篩選效果更好的結論,而對于使用總體RNA表達矩陣篩選差異lncRNA的FPR更高,這一現(xiàn)象背后的統(tǒng)計學原理缺乏深入的剖析。但通過我們對mRNA 和lncRNA 表達水平分布的統(tǒng)計可以看出,二者顯然具有不同的分布規(guī)律,這或許是導致這種現(xiàn)象的關鍵原因。由于DESeq2軟件包采用的標準化模型較為復雜,仍待后續(xù)進一步的數理驗證。此外,本研究分析所采用的樣本量較少,尤其在組內分析時較難得到有意義的結果,之后我們將繼續(xù)在樣本量更大的數據集中進一步研究和驗證。綜上,本研究通過對含有ERCC spike-in對照的全轉錄組測序數據進行差異分析,發(fā)現(xiàn)直接使用lncRNA表達矩陣篩選差異lncRNA的方法特異性和準確性更好。這為今后探究多樣化的差異lncRNA篩選方法提供了一定的理論依據。
利益沖突聲明/Conflict of Interests
所有作者聲明不存在利益沖突。
All authors disclose no relevant conflict of interests.
作者貢獻/Authors'Contributions
魏豪主要完成數據分析處理與文章撰寫的工作,邱家俊主要負責數據集檢索與數據分析指導,顏景斌主要負責總體研究思路和論文整體構思。所有作者均閱讀并同意了最終稿件的提交。
WEI Hao completed the work of data analysis and wrote the manuscript. QIU Jiajun completed the dataset retrieval and guided the data analysis. YAN Jingbin designed and supervised the overall research ideas and the overall conception of the paper.All the authors have read the last version of paper and consented for submission.
·Received:2022-03-24
·Accepted:2022-07-14
·Published online:2022-07-28
猜你喜歡
音樂探索(2022年2期)2022-05-30 21:01:37
美與時代·美術學刊(2022年3期)2022-04-27 01:18:15
口腔護理用品工業(yè)(2021年4期)2021-11-02 08:22:56
小天使·一年級語數英綜合(2019年8期)2019-08-27 02:23:00
人大建設(2019年12期)2019-05-21 02:55:32
小學科學(學生版)(2018年7期)2018-08-13 09:33:04
中國公路(2017年9期)2017-07-25 13:26:38
汽車維修與保養(yǎng)(2015年8期)2015-04-17 03:32:51
鄭州大學學報(醫(yī)學版)(2015年2期)2015-02-27 14:50:46
中國質量與標準導報(2014年9期)2014-02-28 22:25:45
