穩定的釕基離子液體催化液相乙炔氫氯化
2022-09-20 08:18:06姚世康張旭斌王富民任雁飛
化學工業與工程 2022年4期
關鍵詞:催化劑
姚世康張旭斌王富民任雁飛
(天津大學化工學院,天津 300350)
聚氯乙烯(Polyvinyl Chloride,PVC)是五大通用塑料之一,氯乙烯單體是生產聚氯乙烯的主要原料,具有不飽和雙鍵結構,可以與多種單體發生共聚,最終被加工成各種管材、薄膜、建材等實用化工產品[1,2]。 其生產工藝主要有乙烯氧氯法、平衡氧氯法和電石乙炔法[3]。 我國工業生產氯乙烯采用以煤炭為原料的乙炔法,目前所用的HgCl2易揮發,危害生態環境和人類健康,因此,亟需開發一種新型的非汞催化劑[4]。
20 世紀60~90 年代,Smith、Hutchings 及Shinoda 等[5-8]通過實驗,將催化劑活性與金屬離子電子親和勢相關聯,驗證了AuCl3催化劑的初始催化活性遠高于HgCl2和PdCl2。 與此同時,他們發現AuCl3催化劑在反應過程中失活較快,催化穩定性較差。
Xu 等[9]通過DFT 計算,探討了Au 催化體系引入離子液體后,催化反應的路徑和機理。 李小年課題組[10]采用負載的方式制備了Au-IL/AC 催化劑,在溫度180 ℃、乙炔空速370 h-1、HCl 與C2H2氣速比為1.2 時,乙炔轉化率可達72%。 Zhao 等[11]制備了Au-Cu-IL/AC 催化劑,在170 ℃、乙炔空速740 h-1、HCl 與C2H2氣速比為1.2 時,乙炔轉化率可達76.7%,明顯高于Au-IL/AC。
近年來,金屬釕(Ru)因具有高活性、低價格和環境友好的特點,有望成為無汞催化體系的核心。張金利課題組[12]制備了1%Ru@ 15%TPPB/AC(質量比,下同)催化劑,在溫度為170 ℃、乙炔空速為360 h-1的條件下在48 h 內,乙炔轉化率可達99.7%。
2018 年,李航等[13]以氯化膽堿為配體,制備了0.2%Ru-10%ChCl/AC(wt)催化劑,在溫度170 ℃、乙炔空速900 h-1的條件下,反應進行25 h,乙炔轉化率可達87.5%,氯乙烯選擇性超過99.3%,表征分析ChCl 的加入使反應過程中高價態的Run+含量增多。
與氣固相反應相比,氣液相反應具有溫度均一、傳質優異的特點。 不易揮發,溶解能力強的離子液體脫穎而出。 Hu 等[14]將金屬納米粒子(NPs)束縛在陰離子表面活性離子液體(ASC-ILs)中,得到NPs/ASC-ILs 催化劑。 在溫度180 ℃,濃度為0.038 mol·L-1時,Pd-NPs/[P4444][C17COO]具有最佳催化效果,乙炔轉化率可達93%。 隨后,為了降低體系黏度,該小組[15]加入正十四烷作為稀釋劑,得到兩相體系的催化劑,在由下而上的氣流帶動下,NPs@ IL 液滴成為獨立的微反應器單元。
Zhou 等[16]采用1-烷基-3-甲基咪唑類無金屬離子液體催化劑,研究了陰、陽離子種類對催化性能的影響。 當使用[Bmim]Br 時,反應初期溴乙烯是主要的反應產物,隨著反應繼續,溴乙烯逐漸減少而氯乙烯成為主要產物,證明離子液體的陰離子參與反應,經理論模擬得出液相催化體系中N 雜環類離子液體的陰離子以Cl-離子為最適宜。
2019 年,任雁飛等[17]制得N-甲基吡咯烷酮鹽酸鹽,與CuCl 合成制備得到氯亞銅酸離子液體應用于乙炔氫氯化液相反應,在溫度180 ℃、乙炔空速50 h-1、HCl/C2H2氣速比為1.2 的條件下,乙炔轉化率達到86%。
基于上述研究指導,本研究制備了N-甲基吡咯烷酮鹽酸鹽([Hnmpo]Cl)為液相乙炔氫氯化催化反應溶劑,以Ru 為催化活性中心,制備了氯釕酸離子液體,測試了催化性能,隨后加入Sn 構成雙金屬催化體系,在最優反應條件下評價了催化性能,并針對積碳、穩定性以及活性組分價態變化做了表征分析和理論計算,研究了影響其穩定性的因素,從分子角度揭示了HCl 和C2H2在離子液體上的吸附行為,并提出了循環催化反應機理。
1 實驗部分
1.1 試劑與儀器
三氯化釕(RuCl3),氯化亞錫(SnCl2),1-丁基-3甲基咪唑氯鹽([Bmim]Cl),1-己基-3 甲基咪唑氯鹽([Hmim]Cl),四丁基氯化磷([P4444]Cl),四丁基氯化銨([N4444]Cl),皆為分析純,上海邁瑞爾化學品有限公司;N-甲基吡咯烷酮(NMP),分析純,上海阿拉丁生化科技股份有限公司;無水乙醇(C2H5OH),乙酸乙酯(C2H5OOCCH3),蒸餾水(H2O),氫氧化鈉(NaOH),分析純,天津市元立化工技術有限公司;高純氮氣(N2),體積分數為99.999%,高純氫氣(H2),體積分數為99.999%,天津環宇氣體有限責任公司;氯化氫(HCl),體積分數為99.9%,乙炔(C2H2),體積分數為99.9%,東祥特種氣體有限公司。
質量流量計,S4932/MT,北京堀場匯博隆精密儀器有限公司;色譜工作站,Vostro 270-R 586,戴爾(中國)有限公司;氣相色譜儀,3420 A,北京北分瑞利分析儀器有限公司。
1.2 離子液體催化劑的制備
1.2.1 [Hnmpo]Cl 的制備
將100 mL(物質的量1. 037 mol)的N-甲基吡咯烷酮(NMP)加入燒瓶中,攪拌加熱到60 ℃,持續通入純凈氮氣排凈空氣,1 h 后升溫至120 ℃,用質量流量計控制氯化氫流速為21. 5 mL·min-1,設置NaOH 溶液為尾氣吸收,持續20 h 之后停止通氣,將體系密閉120 ℃恒溫攪拌12 h。 將瓶中液體用乙酸乙酯洗滌抽濾共3 次,以除去未反應的原料,得到白色晶體,在60 ℃下真空干燥12 h,即可得[Hnmpo]Cl。
測量制得[Hnmpo]Cl 的1H NMR 以確定其純度。 N-甲基吡咯烷酮鹽酸鹽[Hnmpo]Cl:1H NMR(400 MHz, DMSO-d6,δppm)1.83~1.97(m, 2H,NCH2), 2.18(t, 2H, CH2), 2.69(d, 3H,NCH3), 3.30(td, 2H, COCH2), 13.20(s, 1H,NH)。
1.2.2 離子液體催化劑的制備
在氮氣的保護下,將[Hnmpo]Cl 置于燒瓶中,在120 ℃下把RuCl3一次性加入到燒瓶中,恒溫混合攪拌24 h,即可得到[Hnmpo]Cl-RuCl3IL。
RuCl3-SnCl2/[Hnmpo]Cl IL 的制備方法與之相同。 使用x,y表示離子液體的組成,x=n(RuCl3)/V(IL),y=n(SnCl2)/V(IL),單位為mol·L-1,表示為xRuCl3-ySnCl2/[Hnmpo]Cl IL。
1.3 催化劑催化活性的評價
使用如圖1 所示的裝置測試了離子液體對乙炔氫氯化反應的催化性能。
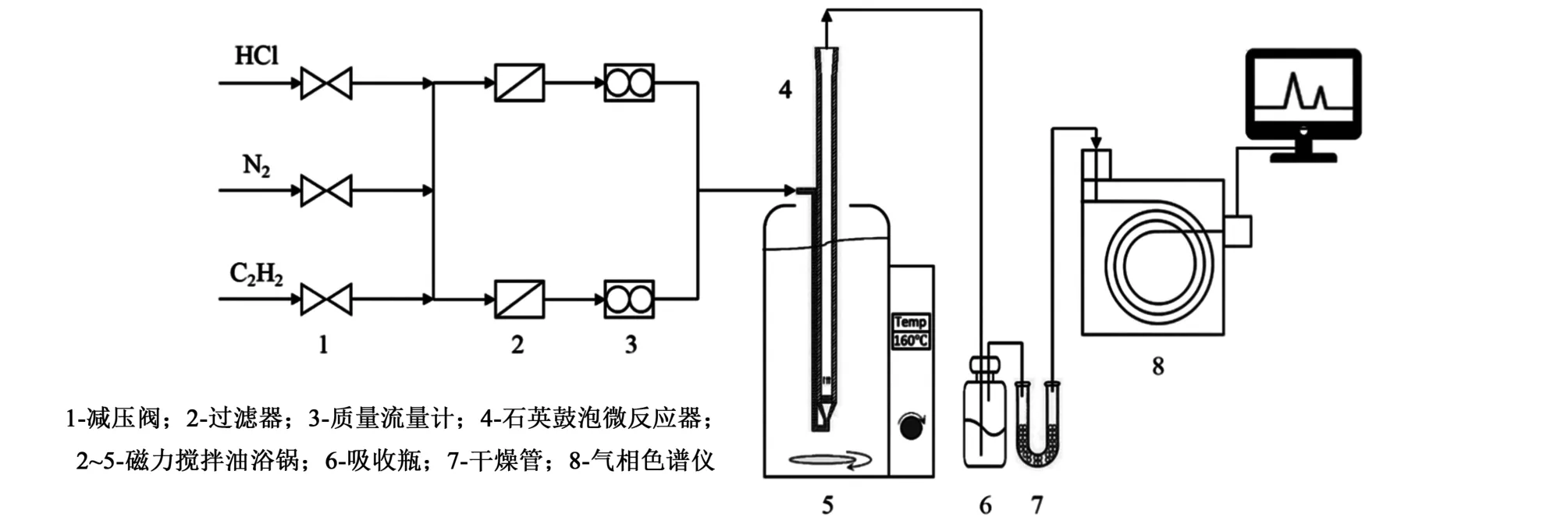
圖1 催化活性評價裝置Fig.1 Experimental setup for catalyst testing
反應氣體氯化氫和乙炔分別由無水氯化鈣和變色硅膠干燥,然后通過過濾器,由經皂膜流量計校正過的質量流量計控制流量,混合后進入反應管。 反應管內裝有12 mL 離子液體,并在反應前使用氮氣吹掃除去水和空氣,然后通入氯化氫30 min,隨后再將乙炔通過反應管。 一般操作條件下,氯化氫氣態流量為11.5 mL·min-1,乙炔氣態流量為10 mL·min-1,HCl/C2H2氣速比為1.15,此時的乙炔空速為50 h-1。 氣體在管內反應之后,管口上方出氣首先經過裝有氫氧化鈉溶液的尾氣吸收瓶以除去未反應的氯化氫,氣流隨后通過裝有無水氯化鈣顆粒的U 型管脫水,最后經過四通閥進入到氣相色譜儀進行在線分析檢測。 氣相色譜儀使用氫火焰離子化檢測器,色譜柱為2 m ×Φ4 mm 的GDX-301填充柱,色譜柱溫度是100 ℃,氣化室溫度是120 ℃,檢測器溫度是150 ℃,使用高純氮氣為載氣,氮氣、氫氣和空氣的氣速分別為30、30 和300 mL·min-1。
1.4 計算方法
通過氣相色譜結果,使用面積歸一法計算乙炔轉化率(X)和氯乙烯選擇性(S),考慮到反應后氯化氫被氫氧化鈉溶液吸收,在反應過程中體系的體積可以認為是恒定的,計算方程式如式(1)和(2):

(1)和(2)中:φA為產物中剩余乙炔體積分數;φVCM為氯乙烯體積分數。
2 結果與討論
2.1 工藝條件
2.1.1 陽離子種類的影響
結合之前的研究考慮到以不同氮雜環、磷受體為陽離子的鹽酸鹽均可與金屬氯化物形成具有高催化活性的離子液體催化劑,出于對HCl 分子活化機理的考慮,統一選擇陽離子的鹽酸鹽即[Bmim]Cl、[Hmim]Cl、[P4444]Cl、[N4444]Cl 和[Hnmpo]Cl分別與0.007 5 mol·L-1的RuCl3制備成離子液體催化劑,在乙炔空速為40 h-1、溫度為160 ℃、HCl/C2H2氣速比為1.15 的條件下測試了 RuCl3摩爾濃度均為0.007 5 mol·L-1的離子液體的催化性能。 評價結果見圖2。
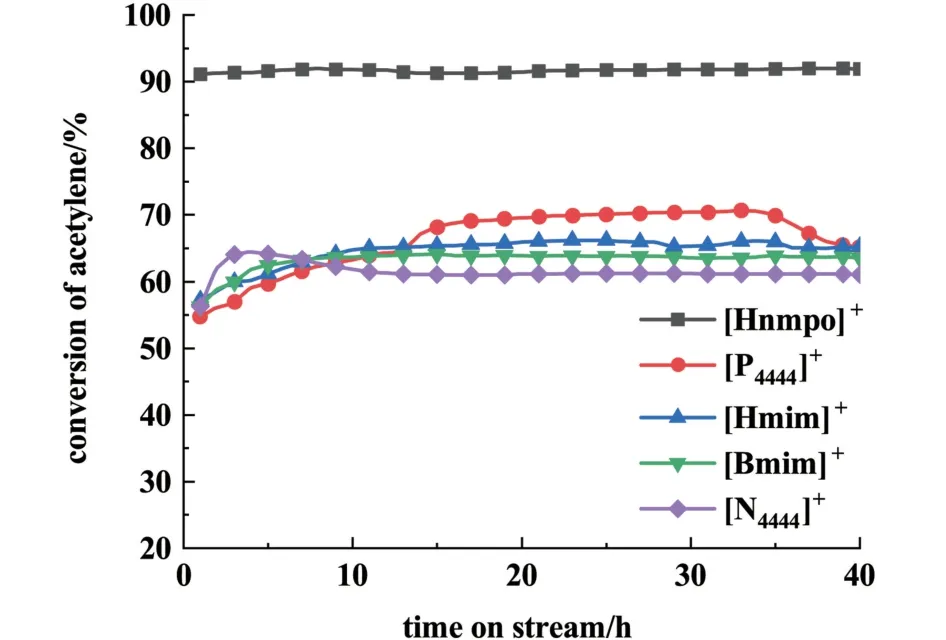
圖2 陽離子種類對催化性能的影響Fig.2 Effect of types of cation rings on catalytic performance
從圖2 中可以看到1-烷基-3 甲基咪唑氯鹽、季膦鹽和季銨鹽分別與RuCl3混合形成的離子液體催化劑的催化性能相差不大,乙炔轉化率不超過70.5%。 [Hnmpo]Cl-RuCl3的催化性能表現最佳,反應25 h 乙炔轉化率為91.3%,而且40 h 內轉化率無下降趨勢,可以認為[Hnmpo]Cl 在以Ru 為活性中心的離子液體體系種有極其重要的作用。
2.1.2 篩選反應條件
隨后,考察了離子液體中RuCl3摩爾濃度、乙炔空速、反應溫度和反應氣體氣速比對反應活性的影響,命名[Hnmpo]Cl-xRuCl3IL,x代表RuCl3的摩爾濃度,單位為mol·L-1,篩選結果見圖3。

圖3 [Hnmpo]Cl-xRuCl3 IL 在不同條件下的催化活性(a)Ru 摩爾濃度的影響;(b)乙炔空速的影響;(c)反應溫度的影響;(d)HCl 與C2H2 氣速比的影響Fig.3 Catalytic activity of [Hnmpo]Cl-xRuCl3 IL under different conditions by the influence of(a)Ru molar concentration;(b)space velocity of acetylene;(c)reaction temperature;(d)the ratio of HCl/C2H2
篩選出最適宜反應條件為RuCl3摩爾濃度0.007 5 mol·L-1、反應溫度170 ℃、50 h-1的乙炔空速以及HCl/C2H2氣速比為1.15。
在此條件下對[Hnmpo]Cl-xRuCl3IL 的壽命進行了測試,結果見圖4(c),乙炔轉化率在140 h 內可保持在89.6%以上,為了進一步提升催化穩定性,考慮對離子液體改性處理。
2.1.3 雙金屬催化體系
結合之前的研究[17]發現,體系內較多的HCl 不僅可以防止[Hnmpo]Cl 的分解,還可以有效避免Ru 因吸附過多的C2H2而被還原,從增強對HCl 的吸附出發,考慮到SnCl2在液態環境中與HCl 有較強的結合能力,因此加入SnCl2構成雙金屬催化體系以增強對HCl 的吸附能力,相關反應見式(3)。

首先使用SnCl2與[Hnmpo] Cl 混合制備了[Hnmpo]Cl-ySnCl2IL,y代表SnCl2的摩爾濃度,單位為mol·L-1,在反應溫度170 ℃、乙炔空速50 h-1、HCl/C2H2氣速比為1.15 的條件下考察了SnCl2單組分的催化活性,結果見圖4(a)。 可以看出,在相同的反應條件下,[Hnmpo]Cl-ySnCl2IL 的催化性能與純[Hnmpo]Cl 的催化性能相差無幾,可以證明Sn物種對催化活性沒有貢獻。
隨后制備了n(Ru)∶n(Sn)分別為1 ∶1,1 ∶2,1 ∶3和1 ∶4的xRuCl3-ySnCl2/[Hnmpo]Cl 離子液體催化劑(x=0.007 5 mol·L-1),測試了其催化性能,結果見圖4(b),可以看出,不加入SnCl2時,反應140 h之后乙炔轉化率開始逐漸下降,當Ru/Sn 物質的量之比為1 ∶3,反應200 h 之內,乙炔轉化率最高可達96.1%,且穩定性良好,所以繼續對其進行壽命測試,結果見圖4(d),可以看出,優化后的雙金屬催化劑在反應700 h 內,乙炔轉化率保持在 96.1%,氯乙烯選擇性保持在99.7%,表現出了極強的穩定性。
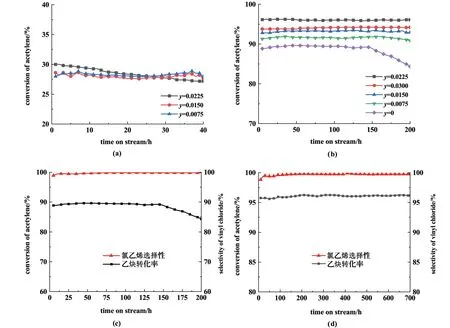
圖4 (a)[Hnmpo]Cl-ySnCl2 IL 的催化活性;(b)金屬摩爾的影響;(c)復配前的壽命;(d)復配后的壽命Fig.4 (a)Catalytic activity of [Hnmpo]Cl-ySnCl2 IL;(b)catalytic performance as a function of the ratio of Ru and Sn;(c)life before compounding;(d)life after compounding
2.2 離子液體的表征
為了探究影響催化反應活性和催化穩定性的因素,對單金屬離子液體和雙金屬離子液體做了一系列表征分析(2.2 節中所有離子液體催化劑,x=0.007 5,y=0.022 5, 單位均為mol·L-1)。
2.2.1 離子液體的陰、陽離子結構
為了確認陰、陽離子的具體形態,分別研究了[Hnmpo]Cl-xRuCl3在正負離子模式下的高分辨質譜(ESI-MS),結果見圖5。
從圖5(a)可以看出,[Hnmpo]Cl-xRuCl3IL 中存在N-甲基吡咯烷酮的陽離子[Hnmpo]+,其中,正離子模式分子離子峰的質荷比m/z為100.131 1,理論質荷比為100.131 0,表征結果與理論值是一致的。 圖5(b)顯示[Hnmpo] Cl-xRuCl3IL 中存在RuCl-4離子,其中,負離子模式分子離子峰的質荷比m/z分別為241.733 2、243.774 1 和245.770 3,分別對應3 種不同同位素組合下的和。 RuCl-4的理論質荷比為242.884 8,表征結果與理論值是一致的。

圖5 [Hnmpo]Cl-xRuCl3 IL 的電噴霧質譜圖(a)正離子模式;(b)負離子模式Fig.5 ESI-MS spectra of [Hnmpo]Cl-xRuCl3 IL(a)positive ion mode;(b)negative ion mode
2.2.2 離子液體的積碳量
由乙炔、氯乙烯或反應中生成的低聚物聚合所引起的積碳,是乙炔氫氯化反應中固體催化劑失活的常見原因之一。 為了測量反應過程中生成的焦炭含量,在空氣氣氛下使用TGA 對反應前后的[Hnmpo]Cl-xRuCl3IL 進行了測試。 圖6 顯示了新鮮的和在溫度170 ℃、50 h-1乙炔空速下反應48 h后的離子液體的TGA 結果。

圖6 新鮮和反應后的[Hnmpo]Cl-xRuCl3 IL 在空氣氛圍下的TGA 曲線Fig.6 TGA curves of the fresh and used[Hnmpo]Cl-xRuCl3 IL in air atmosphere
反應中生成的焦炭所引起的失量主要在250~400 ℃溫度范圍內[18]。 可以計算出,反應過程中積碳量僅為0.51%,可以認為[Hnmpo]Cl-xRuCl3IL在液相體系催化乙炔氫氯化反應能夠較好抑制積碳生成,展現出均相體系的最明顯優勢。
2.2.3 離子液體的分子間作用力
研究了[Hnmpo]Cl 和[Hnmpo]Cl-xRuCl3IL 的FTIR 譜,結果見圖7。
從圖7 中可以看出,[Hnmpo]Cl 與RuCl3形成[Hnmpo]Cl-xRuCl3IL 之后,位于3 396.0 cm-1處的N—H 伸縮振動峰向更高的波數移動了14.5 cm-1,烷基C—H 伸縮振動峰都發生紅移至更低的波數,伸縮振動峰從1 650.3 cm-1紅移至1 641.6 cm-1。 這些特征峰的變化都表明了[Hnmpo] ClxRuCl3IL 中的NMP 陽離子與含Ru 陰離子之間確實存在較強的相互作用力, 可能主要歸結于C—H…Cl 氫鍵作用力。

圖7 [Hnmpo]Cl-xRuCl3 IL 和[Hnmpo]Cl 的 FTIR 譜圖Fig.7 FTIR spectra of [Hnmpo]Cl-xRuCl3 IL and[Hnmpo]Cl
為了從分子與化學鍵的角度揭示離子液體的陰、陽離子的作用方式,我們計算了[Hnmpo]Cl-xRuCl3IL 的靜電勢(Electrostatic potential)、自然布局分析(Natural population analysis, NPA)以及HCl 分子的吸附結構。 所有理論計算結果如圖8 和圖9 所示。
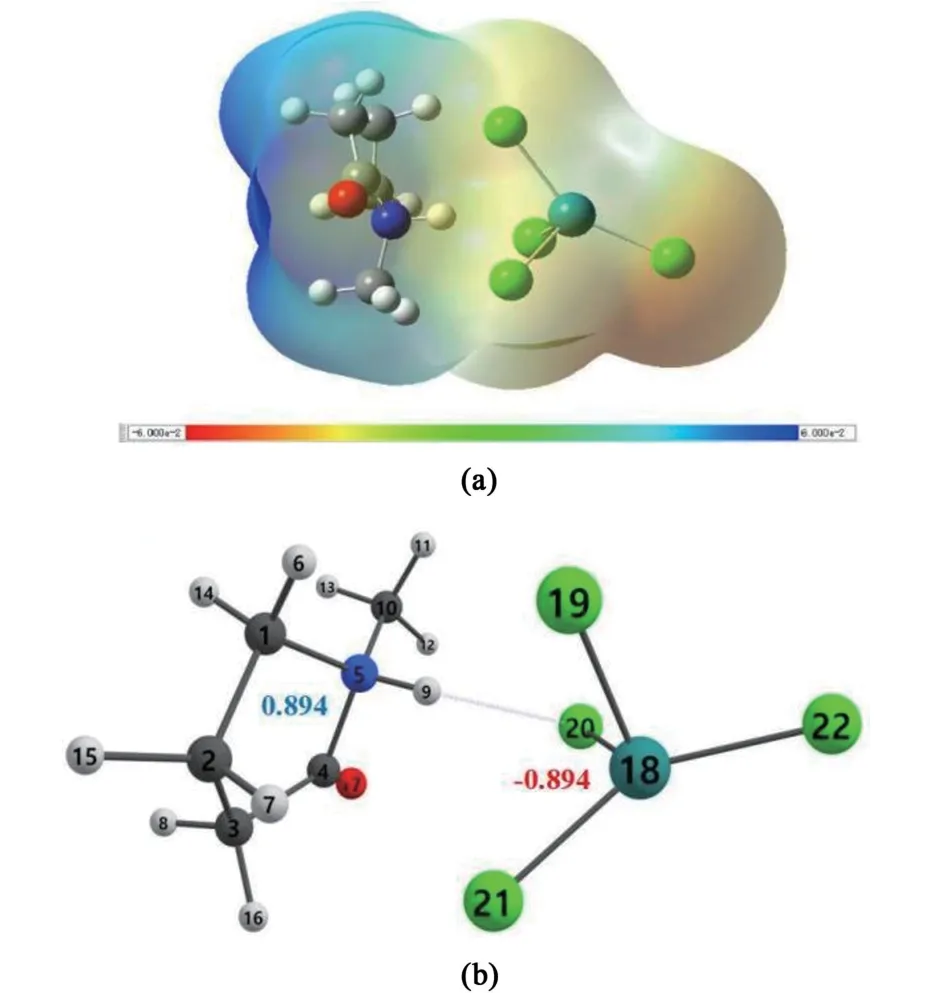
圖8 新鮮[Hnmpo]Cl-xRuCl3 IL 的(a)靜電勢;(b)分子間氫鍵, IL+與IL-的NPA 電荷分別用藍色(正電)和紅色(負電)數字表示Fig.8 (a)Electrostatic potential and(b)intermolecular hydrogen-bonding of fresh [Hnmpo]Cl-xRuCl3 IL with IL+and IL-NPA charges represented in blue(e+)and red(e-)

圖9 HCl 分子吸附在[Hnmpo]Cl-xRuCl3 IL 上的結構Fig.9 Structure of [Hnmpo]Cl-xRuCl3 IL adsorbing HCl
圖8(a)顯示,[Hnmpo]Cl-xRuCl3IL 形成了2個明確的帶電基團,RuCl3與[Hnmpo]Cl 結合后,由于Cl 原子的配位作用,形成了極性更大的RuCl-4陰離子,成為體系里的負電荷中心,在催化反應中提供親核位點,同時,親電位點Ru 被這些親核配體環繞,這將更有利于Ru 對C2H2分子的吸附作用。
此外,圖8(b)說明2 個帶電基團并不是傳統意義上溶液內的陰、陽離子,正、負電基團將一部分電荷離域,貢獻給分子間作用力,表現為[Hnmpo]+與RuCl-4之間的H…Cl 氫鍵,即電荷輔助氫鍵(Chargeassisted hedrogen bond, CAHB)[24],并且CAHB 的鍵合強度相當于共價鍵,這說明H…Cl 增強了離子液體的熱穩定性。
2.2.4 離子液體的熱穩定性
對新鮮的[Hnmpo]Cl-xRuCl3IL 作了熱重-紅外光譜聯用分析(TG-IR),研究其分解溫度及對應分解產物,結果見圖10 和圖11。

圖10 新鮮[Hnmpo]Cl-xRuCl3 IL 在氮氣氣氛下的TGA-DTG 曲線Fig.10 TGA-DTG curves of fresh [Hnmpo]Cl-xRuCl3 IL in N2 atmosphere
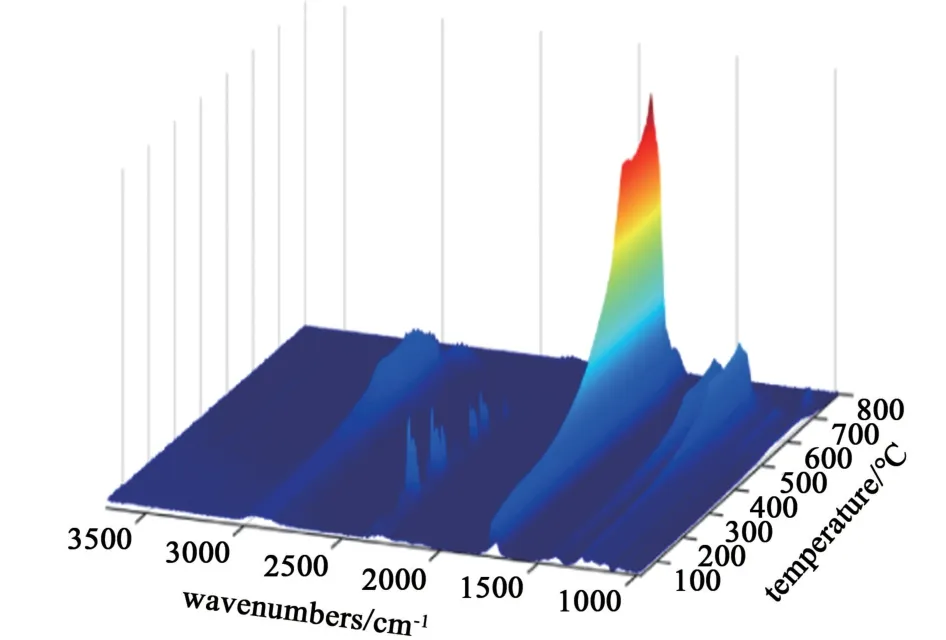
圖11 [Hnmpo]Cl-xRuCl3 IL 在TGA 升溫過程中得到的同步三維FTIR 譜圖Fig.11 3D FTIR spectra of [Hnmpo]Cl-xRuCl3 IL with TGA heating
結合圖10 的2 條曲線可以看出,離子液體在升溫過程中有2 個不同的失量過程。 首先是100~164.1 ℃,對應于占離子液體絕大比重的游離[Hnmpo]Cl 受熱分解揮發,164~288 ℃的失量過程則主要是與極低濃度的RuCl3結合形成離子液體的極少部分[Hnmpo]Cl 的分解揮發。 這2 個過程分界明顯,證明[Hnmpo]Cl-xRuCl3IL 中確實存在與計算結果相符的分子間氫鍵。
從圖11 可以清晰地看出不同溫度下的特征峰位置完全一致,處于3 010.1~2 807.6 cm-1內的寬峰對應HCl 分子的H—Cl 伸縮振動,1 736.1 cm-1處的強峰則因NMP 分子的伸縮振動引起[25],1 308.2~1 389.2 cm-1的2 處峰可歸因于NMP 分子的C—N 伸縮振動,說明分解產物只有NMP 和HCl 分子,證明離子液體在持續通入HCl 的環境下是熱穩定的。
使用ICP-OES 測量了新鮮和反應36 h 之后的xRuCl3-ySnCl2/[Hnmpo]Cl IL 中Ru 元素含量,結果如表1 所示,可以看出,反應前后離子液體中的Ru含量與標準值都非常接近,證明活性物種在反應過程中并無損失。

表1 新鮮和反應后的xRuCl3-ySnCl2/[Hnmpo]Cl IL 中的Ru 含量Table 1 Ru content in the fresh and used xRuCl3-ySnCl2/[Hnmpo]Cl IL calculated from ICP-OES
通過XPS 測量新鮮和反應后的雙金屬離子液體,得到的全譜圖和表面化學元素組成分別顯示于表2 和圖12 中。
如圖12 所示,反應前后的XPS 全譜圖曲線基本相同。 由表2 可以得到,反應前后Ru/N 原子比分別為0.071 和0.072,基本沒有變化,結合ICP-OES中Ru 含量的數據,證明xRuCl3-ySnCl2/[Hnmpo]Cl IL在反應過程中可以保持穩定,活性組分與[Hnmpo]Cl 幾乎無損失。
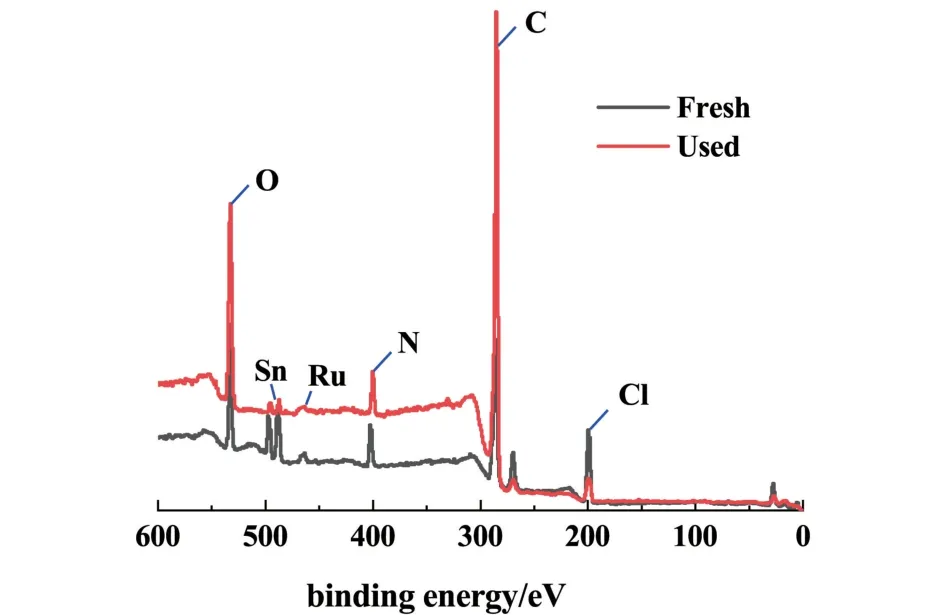
圖12 新鮮和反應后xRuCl3-ySnCl2/[Hnmpo]Cl IL 的XPS 全譜圖Fig.12 XPS sweep scan of the fresh and the used xRuCl3-ySnCl2/[Hnmpo]Cl IL

表2 新鮮和反應后的xRuCl3-ySnCl2/[Hnmpo]Cl IL表面化學元素組成Table 2 The elemental composition of the fresh and used xRuCl3-ySnCl2/[Hnmpo]Cl IL from XPS
2.2.5 離子液體中的Ru 物種價態
乙炔氫氯化反應過程中金屬活性組分發生價態變化也是催化劑失活的主要原因之一[19,20],因此使用XPS 表征分析了新鮮和反應后的xRuCl3-ySnCl2/[Hnmpo]Cl IL 中Ru 的價態變化。
首先,在XPS 分析中選用Ru 3p 軌道來分析,這是由于Ru 3d 軌道的結合能與C 1s 軌道的結合能會發生重合而對分析造成干擾[21]。 因此,為了得到清晰的卷積結果,采用信號較強的Ru 3p3/2。 圖13 與表3 分別為Ru 3p3/2峰的去卷積譜圖和Ru 物種的相對含量。

表3 反應前后的[Hnmpo]Cl-xRuCl3 IL 和xRuCl3-ySnCl2/[Hnmpo]Cl IL 中Ru 物種相對含量Table 3 The relative contents of ruthenium species inthe fresh and used [Hnmpo]Cl-xRuCl3 IL and xRuCl3-ySnCl2/[Hnmpo]Cl IL from XPS
從圖13 可以看出,反應前后離子液體催化劑樣品的Ru 含3 個價態,結合文獻報道[12,21,22],液態催化劑中Ru 物種主要存在3 種類型,其分別為:Run+(1≤n<3)[462.3(±0.3)eV]、Ru3+[463.5(±0.3)eV]、Ru4+[464.8(±0.2)eV]。

圖13 新鮮和反應后xRuCl3-ySnCl2/[Hnmpo]Cl IL 的Ru 3p3/2 XPS 圖譜(a)fresh;(b)usedFig.13 Deconvolution profiles of Ru 3p3/2 XPS spectrum of the fresh and used xRuCl3-ySnCl2/[Hnmpo]Cl IL(a)fresh;(b)used
表3 顯示,xRuCl3-ySnCl2/[Hnmpo]Cl IL 在反應過程中,高價態的Ru 物種Ru3+和Ru4+被還原成低價Run+的比例大大減小,據文獻報道[21,22],高價態Ru 含量是活性與穩定性的決定性因素,說明Sn的加入有效抑制了高價態Ru 的還原,延長了催化劑壽命。
2.2.6 Sn 金屬復配的影響
采用UV-Vis 檢測離子液體中可能存在的配合物。
圖14 為新鮮和反應36 h 后,離子液體的紫外可見吸收光譜。 可以發現,反應前后2 種離子液體在222 nm 左右都出現了強吸收峰,這對應于N-甲基吡咯烷酮[26]和Ru3+與[Hnmpo]Cl 形成的配合物[27,28]。 除了新鮮的[Hnmpo]Cl-RuCl3IL,其它3條曲線都在264 nm 附近出現了1 個很弱的吸收峰,更明顯的是,新鮮的xRuCl3-ySnCl2/[Hnmpo]Cl IL在264 nm 附近同樣存在吸收峰,說明在反應前,SnCl2的加入就可以使Ru 形成新配合物。

圖14 新鮮和反應后[Hnmpo]Cl-xSnCl2 IL 和xRuCl3-ySnCl2/[Hnmpo]Cl IL 的UV-Vis 曲線Fig.14 UV-Vis spectra of the [Hnmpo]Cl-xSnCl2 IL and xRuCl3-ySnCl2/[Hnmpo]Cl IL
為了研究加入Sn 組分后,對離子液體陰、陽離子基團帶電量和分子間作用力的影響,計算了新鮮xRuCl3-ySnCl2/[Hnmpo]Cl IL 的靜電勢和自然布局分析(NPA),結果如圖15 所示。
圖15(a)顯示,SnCl2的引入給Ru 提供了新的Cl 原子配位,形成[RuCl3-(Cl)2-SnCl]-陰離子。 圖15(b)顯示,雙金屬離子液體的NPA 電荷更小,說明H…Cl 更強,分子間共價作用力更大使體系更穩定。
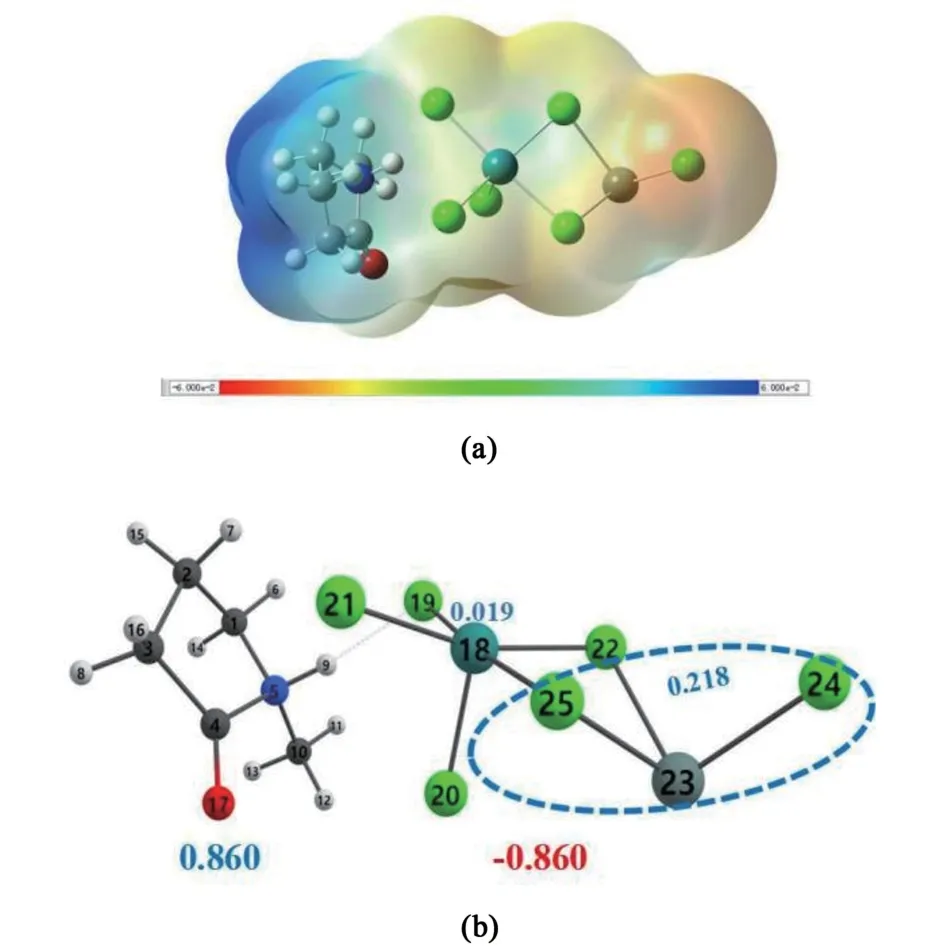
圖15 新鮮xRuCl3-ySnCl2/[Hnmpo]Cl IL 的(a)靜電勢;(b)分子間氫鍵, IL+與IL-的NPA 電荷分別用藍色(正電)和紅色(負電)數字表示Fig.15 (a)Electrostatic potential and(b)intermolecular hydrogen-bonding of fresh xRuCl3-ySnCl2/[Hnmpo]Cl IL with IL+and IL-NPA charges represented in blue(e+)and red(e-)
2.2.7 氣體分子在離子液體上的吸附行為
隨后,我們計算了2 種離子液體吸附HCl 和C2H2時的吸附能以及相應結構的分子軌道能量。
HCl 和C2H2在[Hnmpo]Cl-xRuCl3上的吸附能ΔE分別為-56.85 和-82.76 kJ·mol-1,證明離子液體對HCl 和C2H2有很強的吸附能力,與較高的催化活性相符。 加入SnCl2后,主要增強了離子液體對HCl 的吸附能力,提升了HCl 占據催化劑表面活性位點的競爭力,揭示了催化穩定性得到顯著提升的內在規律。

表4 HCl 和C2H2 分別在離子液體上的吸附能ΔETable 4 The calculated adsorption energy(ΔE)of HCl and C2H2 on ILs respectively
前線軌道是化學反應中最活躍的軌道,能量差代表電子躍遷的難易程度。 吸附過程中電子從離子液體向HCl 轉移形成氫鍵,因此HOMO 能量越高表示越易吸附活化HCl,而C2H2通常作為給電子體與金屬原子絡合形成π 鍵被吸附,因此LUMO 能量越低表示越易接受電子吸附和活化C2H2。
表5 顯示,xRuCl3-ySnCl2/[Hnmpo] Cl IL 的HOMO 能量比[Hnmpo]Cl-xRuCl3IL 顯著升高,證明離子液體吸附HCl 的能力得到增強。xRuCl3-ySnCl2/[Hnmpo]Cl_C2H2擁有更高的HOMO 能量,說明其在先吸附C2H2后繼續吸附HCl 分子的能力更強,提高了HCl 的吸附競爭力。 另外,計算出2 種IL_C2H2的LUMO-HOMO 能級差分別為2.729 和3.431 eV,分子內電子躍遷所需的能量顯著增大,同結構的LUMO-HOMO 能級差代表了其發生二聚的能力,數值越小表示越容易發生,因此[Hnmpo]ClxRuCl3_C2H2更容易發生二聚,雙金屬離子液體的能級差較之顯著增大,因此催化活性和穩定性都有所提升,與上述關于吸附能的判斷一致。

表5 離子液體吸附HCl 和C2H2 時的分子軌道能量Table 5 The calculated orbital energy of the ionic liquids absorbing HCl and C2H2 respectively
2.2.8 催化反應機理
將[Hnmpo][RuCl3-(Cl)2-SnCl]作為催化組分模型進行了DFT 計算,主要包括原子行為,電荷轉移和能量變化,優化為循環,催化反應機理如圖16所示。
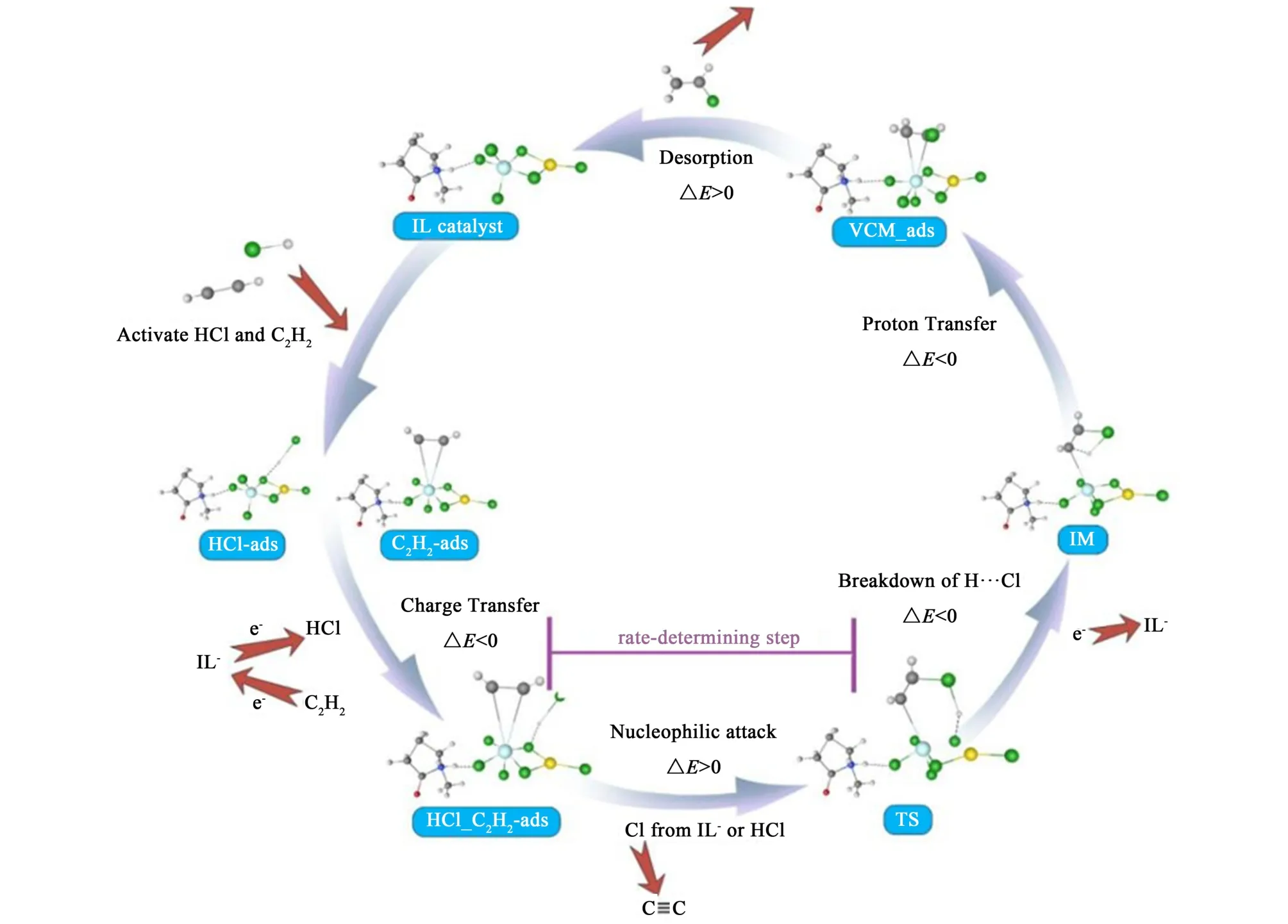
圖16 理論計算xRuCl3-ySnCl2/[Hnmpo]Cl IL 循環催化機理模型Fig.16 Cyclic catalytic mechanism model of the xRuCl3-ySnCl2/[Hnmpo]Cl IL
首先,活性位點受到HCl 和C2H2的進攻,HCl與IL-形成氫鍵Cl…H—Cl 被吸附,本身分子內的H—Cl 鍵被拉長活化,C2H2與Ru 絡合形成π 鍵被吸附,同時暴露出空軌道而被活化,單吸附的結構繼續吸附另一種分子,最終都形成共吸附的HCl_C2H2-ads,此過程e-從IL-轉移至HCl,從C2H2轉移至IL-。 隨后,Cl 原子從IL-中掙脫,形成游離的Cl…H…Cl,因對稱性所致,電子均勻排布,因此對C原子發生親核加成的Cl 原子可以來源于IL-或HCl,此步驟涉及Ru—Cl—Sn 共價鍵的弱化斷裂,需大幅吸熱,故為整個循環中的速率控制步驟,與相關文獻報道一致[17]。 加成后電子向π 鍵離域,H…Cl 極度弱化并斷裂,Cl 重新與IL-配位形成共價鍵,此步驟電子回到IL-,獲得電子的π 電子云密度升高,引發質子轉移形成C—H 鍵,催化過程基本完成,生成吸附態的VCM-ads,最后吸收極少熱量完成C2H3Cl 脫附,電子向IL-轉移,Ru 物種的價態升高,活性位點重生,至此舊循環完成且新循環開始。
3 結論
1)[Hnmpo]Cl-xRuCl3IL 在篩選的最佳反應條件下表現出良好的催化性能。 結合ESI-MS、TGA、FTIR、ICP-OES、XPS 表征和理論計算,確認了陰、陽離子的存在形式,且存在較強的相互作用力,反應前后活性組分幾乎無損失,證明離子液體在反應條件下可以保持穩定。
2)加入 SnCl2復配得到更穩定的xRuCl3-ySnCl2/[Hnmpo]Cl IL 催化劑,在反應700 h 內,乙炔轉化率大于96.1%,氯乙烯選擇性超過99.5%。ICP-OES 和XPS 表征顯示活性物種Ru 在反應前后幾乎無損失,Sn 的加入有效抑制了反應中高價態Ru 的還原,從而抑制了催化劑失活,極大的提升了催化穩定性。
3)對HCl 和C2H2的吸附行為進行DFT 計算后,發現xRuCl3-ySnCl2/[Hnmpo]Cl IL 吸附HCl 和C2H2的能力更強,并且其吸附C2H2的結構LUMOHOMO 能極差較大,不易發生二聚反應。 隨后計算了[Hnmpo][RuCl3-(Cl)2-SnCl]模型催化反應機理,分析了原子行為、電荷轉移和能量變化。
猜你喜歡
大自然探索(2023年7期)2023-11-14 13:08:06
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
智富時代(2018年3期)2018-06-11 16:10:44
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
超硬材料工程(2016年1期)2016-02-28 22:20:04
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
合成化學(2015年4期)2016-01-17 09:01:27
應用化工(2014年3期)2014-08-16 13:23:50
