吡啶甲酰萘胺的C-8位選擇性C-H官能化反應(yīng)研究進展
2022-09-20 07:11:02姜風軒
紹興文理學院學報(自然科學版) 2022年3期
程 凱 姜風軒 蔡 薇 酈 勇
(1. 浙江省精細化學品傳統(tǒng)工藝替代技術(shù)研究實驗室,浙江 紹興 312000;2. 紹興文理學院 化學化工學院,浙江 紹興 312000)
0 引言
過渡金屬催化的C-H鍵官能化已經(jīng)成為有機合成中的重要合成策略,它提供了從簡單起始原料到復雜分子的新途徑,并提高了所需轉(zhuǎn)化的整體效率,從而從根本上改變了應(yīng)對合成挑戰(zhàn)的方式.近幾十年來,以過渡金屬催化作為手段,通過金屬插入激活C-H鍵的領(lǐng)域,已經(jīng)有許多開創(chuàng)性的發(fā)現(xiàn)[1].隨著以C-H鍵作為化學合成起點的研究不斷深入,學者在該領(lǐng)域的探索中發(fā)現(xiàn)C-H鍵斷裂所需要的反應(yīng)能量和位置選擇性的控制是C-H鍵官能化反應(yīng)面臨的兩個主要挑戰(zhàn).在這方面,過渡金屬催化的導向性C-H鍵官能化反應(yīng)顯示出巨大的前景.反應(yīng)的位置選擇性可以通過C-H鍵與活性金屬中心的螯合來控制,并且通過雜原子與過渡金屬催化劑的配位可以顯著促進底物-催化劑的相互作用.
導向性C-H鍵官能化反應(yīng)通常分為兩類,單齒螯合導向的C-H鍵官能化反應(yīng)[2]與雙齒螯合導向的C-H鍵官能化反應(yīng)[3].而雙齒螯合體系中過渡金屬與雙齒導向基團的配位通常被認為是實現(xiàn)C-H鍵活化過程的關(guān)鍵步驟.吡啶甲酰萘胺是一類重要的雙齒螯合底物,可以在過渡金屬(如鈀,銅,鈷等)輔助下發(fā)生區(qū)域選擇性的1-萘胺的C-8位的C-H鍵官能化反應(yīng).如圖1所示,吡啶甲酰萘胺底物通過吡啶和酰胺上的氮原子對過渡金屬進行雙齒螯合作用,實現(xiàn)了對過渡金屬的空間定位,再與空間位置上接近的萘環(huán)上C8-H實現(xiàn)氧化性C-H鍵插入反應(yīng),形成高價過渡金屬絡(luò)合物,并進行進一步催化轉(zhuǎn)化過程.
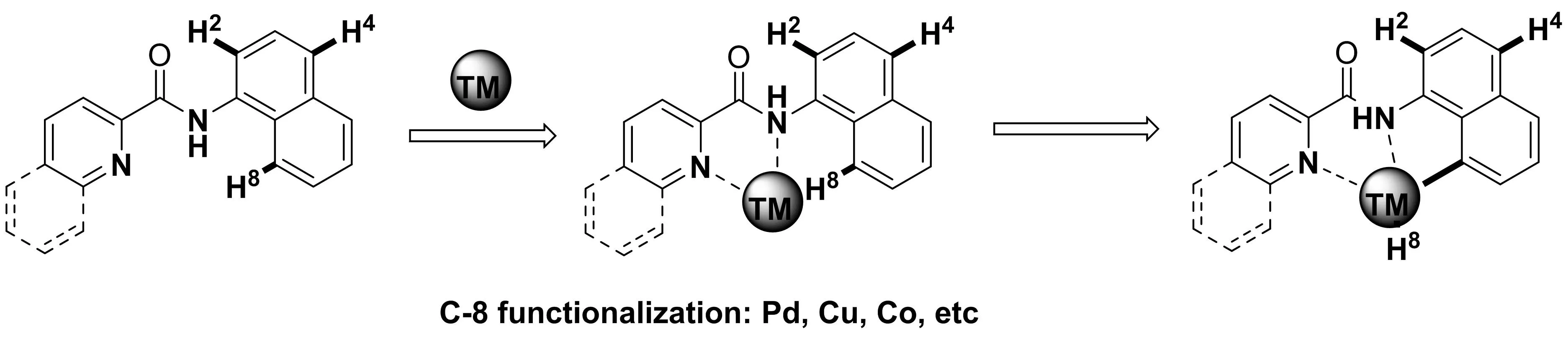
圖1 過渡金屬催化的雙齒導向C-H活化控制的位點選擇性與活化機理
在本文中,以吡啶甲酰萘胺作為研究對象,從不同的過渡金屬催化出發(fā),結(jié)合本課題組在該領(lǐng)域中的研究工作,綜述了雙齒導向基團輔助的C-8位選擇性C-H官能化反應(yīng)的最新研究進展.
1 鈀催化吡啶甲酰萘胺的C-8位選擇性C-H官能化反應(yīng)
2013年,本課題組[4]報道了鈀催化的喹啉/吡啶酰胺導向的1-萘胺與碘代芳烴的C-8芳基化反應(yīng)(圖2).雙齒導向基團(喹啉酰胺)被證明是高度區(qū)域選擇性轉(zhuǎn)化的關(guān)鍵.此外,酰胺導向基團在堿性條件易于水解成一系列8-芳基-1-萘胺衍生物.熱力學計算表明,C-H芳基化反應(yīng)是通過連續(xù)的C-H活化/氧化加成途徑進行的.
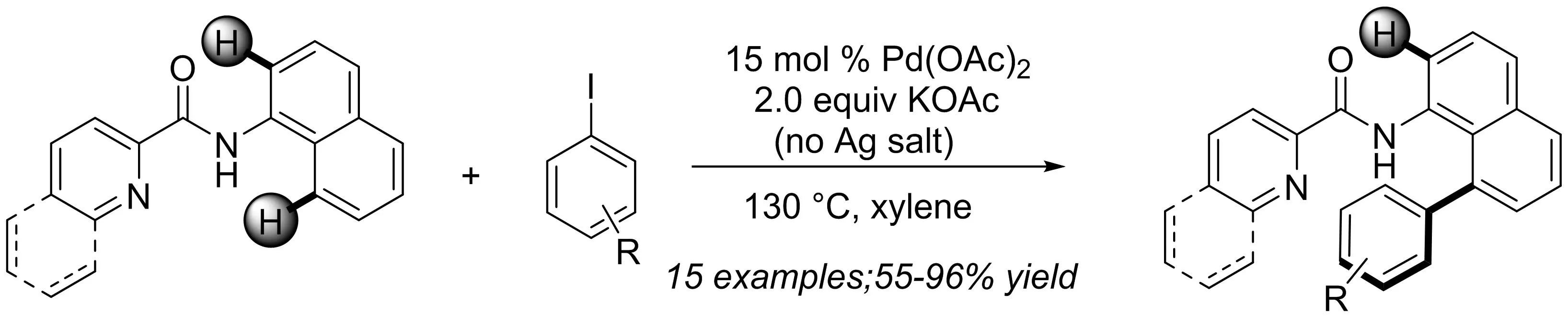
圖2 Pd(II)催化萘酰胺與芳基碘化物的區(qū)域選擇性芳基化反應(yīng)
2014年本課題組[5]又報道了一種鈀催化的含喹啉/吡啶酰胺基團的雙齒導向基團的1-萘胺中C8-H鍵的區(qū)域選擇性烷基化反應(yīng)(圖3),且酰胺導向基團易于在堿性條件下水解掉.各種鹵代烷烴,包括碘代烷烴和芐溴及芐氯化合物可以被用作烷基化試劑,高產(chǎn)率轉(zhuǎn)化為8-烷基-1-萘胺衍生物.

圖3 鈀催化1-萘酰胺與鹵代烷的C8烷基化反應(yīng)
Nishihara課題組[6]報道了一種鈀催化下吡啶酰胺導向的萘胺衍生物的C-H鍵直接硫化反應(yīng)制備8-芳硫基-1-萘胺的簡便途徑(圖4).與傳統(tǒng)的鄰位C-H鍵官能化反應(yīng)不同,該反應(yīng)通過一個五元鈀環(huán)中間體進行,只得到C-8位硫代產(chǎn)物.此外,以二芳基二硒為底物也可以實現(xiàn)相應(yīng)的直接硒化反應(yīng),得到了相應(yīng)的具有良好區(qū)域選擇性的1-萘胺C-8位硒代產(chǎn)物.
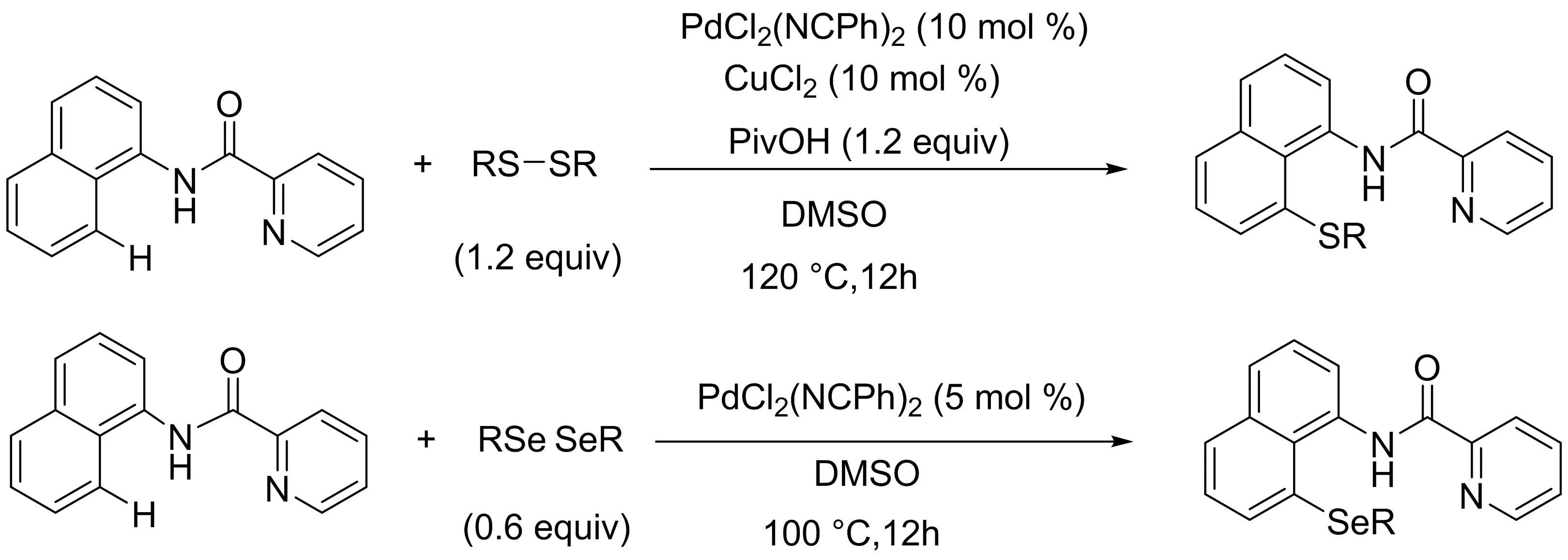
圖4 鈀催化萘胺與二芳基二硫化物的選擇性硫氫化反應(yīng)
以無毒的K4[Fe(CN)6]為氰基化試劑,孫志忠課題組[7]通過鈀催化的C-H鍵活化方法,開發(fā)了一種高效、簡單、環(huán)保的吡啶酰胺導向的直接氰化策略,成功地將一系列N-(1-萘基)吡啶酰胺衍生物轉(zhuǎn)化為相應(yīng)的C-8位直接氰化產(chǎn)物(圖5),還推測了可能的反應(yīng)機理.
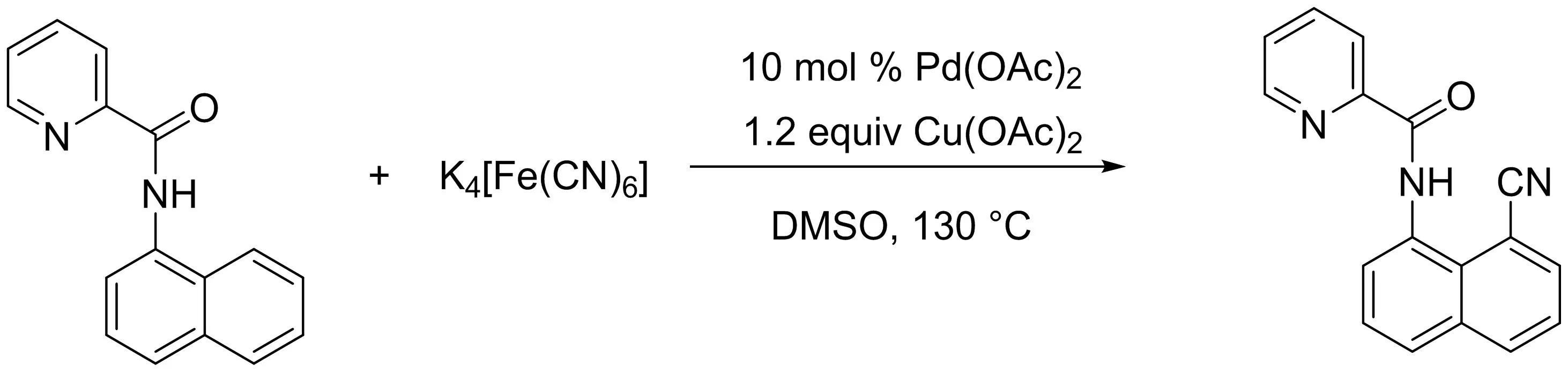
圖5 以K4[Fe(CN)6]為氰化物源的吡啶酰胺直接氰化反應(yīng)
隨后該課題組[8]又報道了雙齒螯合輔助的鈀催化下吡啶酰胺導向的以三甲基氰硅烷為氰基化試劑的C-H鍵直接氰化反應(yīng)(圖6).各種官能團取代的吡啶酰胺衍生物在溫和的條件下以中等到良好的產(chǎn)率反應(yīng)得到相應(yīng)的C-8位氰化產(chǎn)物.此外,氰化產(chǎn)物還可以以較高的產(chǎn)率轉(zhuǎn)化為一些有價值的官能團.
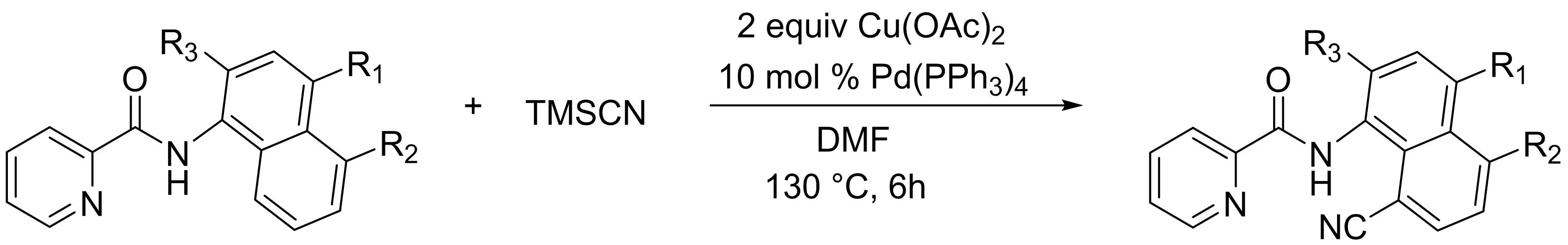
圖6 鈀催化吡啶酰胺雙齒螯合的直接C-H氰化反應(yīng)
吳養(yǎng)潔課題組[9]研究了在鈀催化1-萘胺衍生物與簡單的脂肪族仲胺的C8-H胺化反應(yīng)(圖7),為1,8-萘二胺衍生物的合成提供了一條新途徑.值得注意的是,吡啶酰胺部分作為雙齒導向基團在區(qū)域選擇性轉(zhuǎn)化中發(fā)揮了關(guān)鍵作用.
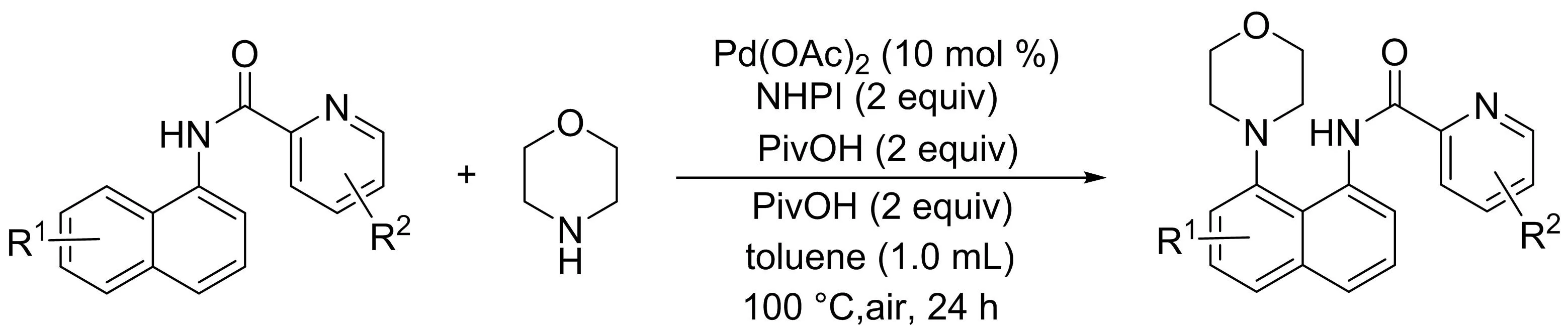
圖7 鈀催化1-萘胺衍生物與脂肪族胺的區(qū)域選擇性C8-H胺化反應(yīng)
隨后該課題組[10]報道了一種簡便、高效的鈀催化1-萘胺衍生物與芳基/烯基酰氯的區(qū)域選擇性C8-H酰化反應(yīng)的方法(圖8).該反應(yīng)具有廣譜官能團耐受性,芳香和α、β不飽和酰氯均能與1-萘胺有效偶聯(lián).此外,吡啶酰胺部分作為一個雙齒導向基團在該區(qū)域選擇性轉(zhuǎn)化中發(fā)揮關(guān)鍵作用.
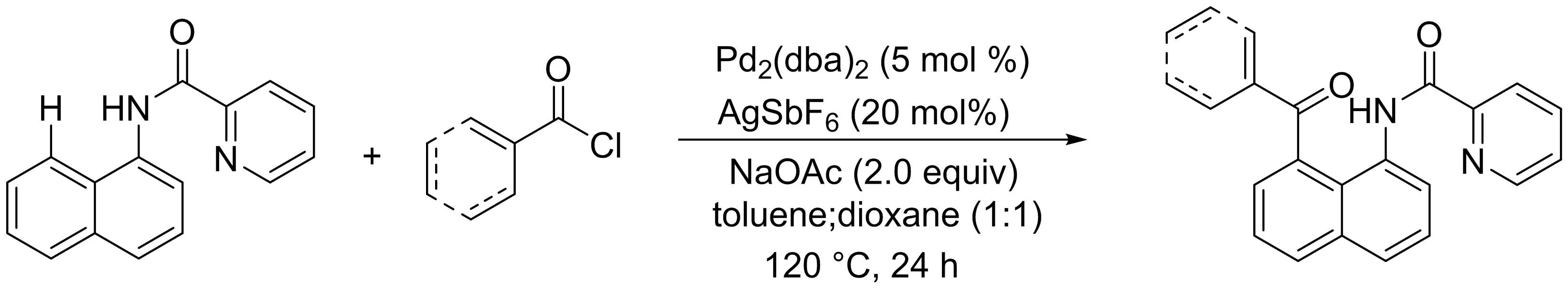
圖8 鈀催化1-萘胺與酰氯的C8-H酰化反應(yīng)
吳養(yǎng)潔課題組[11]研究了1-萘胺衍生物與α-氯代烷基酯在鈀催化下吡啶酰胺導向的C8-H烷氧羰基甲基化反應(yīng)(圖9).該反應(yīng)具有廣闊的底物范圍和良好的官能團耐受性.值得注意的是,α-氯烷基酯作為一種新型的烷基化試劑,可以很容易地通過分子內(nèi)胺化環(huán)化生成內(nèi)酰胺化合物.
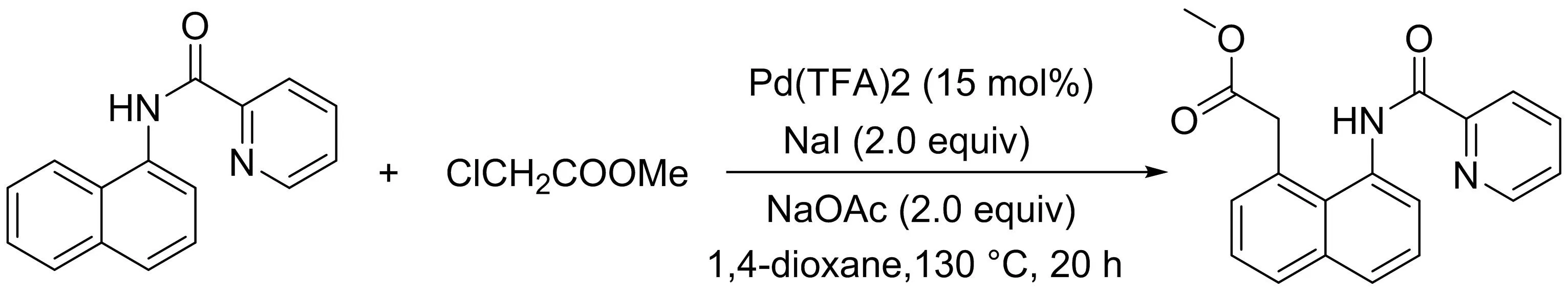
圖9 Pd(II)催化1-萘酰胺與α-氯代烷基酯的C8-H烷氧羰基甲基化反應(yīng)
隨后該課題組[12]又報道了在惰性氛圍下的鈀催化下1-萘胺衍生物與氯甲酸烷基酯的C8-H烷氧羰基化反應(yīng)(圖10),該反應(yīng)具有廣泛的官能團耐受性和高區(qū)域選擇性.此外,該反應(yīng)還具有易于進一步官能化和衍生化的特點.例如,得到的烷氧羰基化產(chǎn)物經(jīng)過多步轉(zhuǎn)化后,合成了一種BET溴蛋白抑制劑-苯[cd]吲哚-2(1H)-酮.控制實驗表明,反應(yīng)可能是經(jīng)歷了自由基歷程,而C-H鍵的裂解不是反應(yīng)的決速步.

圖10 鈀催化1-萘胺與氯甲酸烷基酯的C8-H烷氧羰基化反應(yīng)
本課題組[13]最近報道了以喹啉酰胺為雙齒導向基團的鈀催化1-萘胺與鹵代烷烴的C8-H鍵的直接烷基化反應(yīng)(圖11).各種官能化的鹵代烷烴,包括α-溴代酯和α-溴代酮可以作為偶聯(lián)試劑,區(qū)域選擇性的生成8-烷基-1-萘胺衍生物.而這些烷基化產(chǎn)物的酯基和羰基易于進一步轉(zhuǎn)化為阿啡堿和馬兜鈴生物堿的核心結(jié)構(gòu).
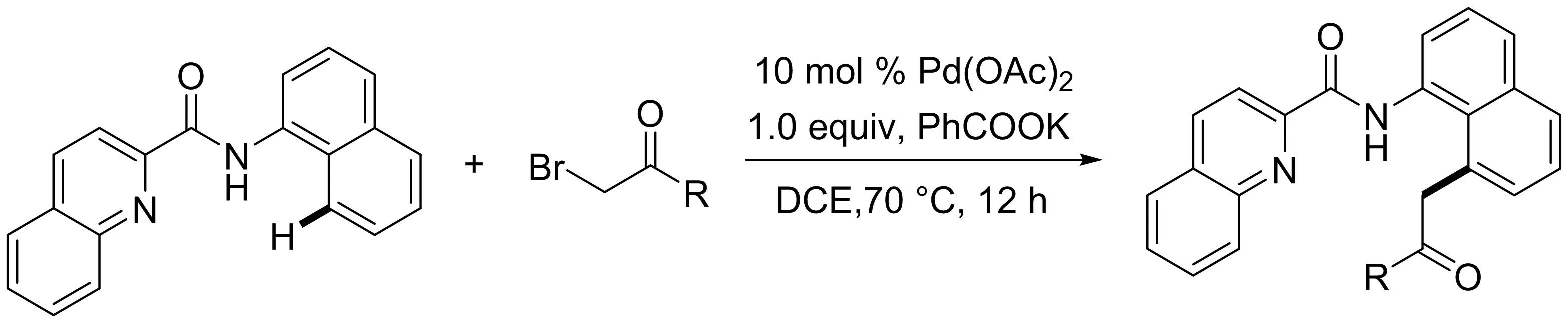
圖11 鈀催化1-萘酰胺的8-烷基化反應(yīng)
Babu課題組[14]報道了鈀催化的雙導向基團輔助的C-H芳基化/烷基化策略在芘結(jié)構(gòu)單元的官能化中的應(yīng)用(圖12).以吡啶酰胺為導向基,鈀催化下的N-(1-芘基)吡啶酰胺的C10位選擇性發(fā)生C-H芳基化/烷基化反應(yīng),得到各種C1,C10-二取代芘,并制備了相應(yīng)的去除導向基團C-H芳基化/烷基化產(chǎn)物.作者通過X射線結(jié)構(gòu)分析確定了其具有代表性的芘衍生物的結(jié)構(gòu).
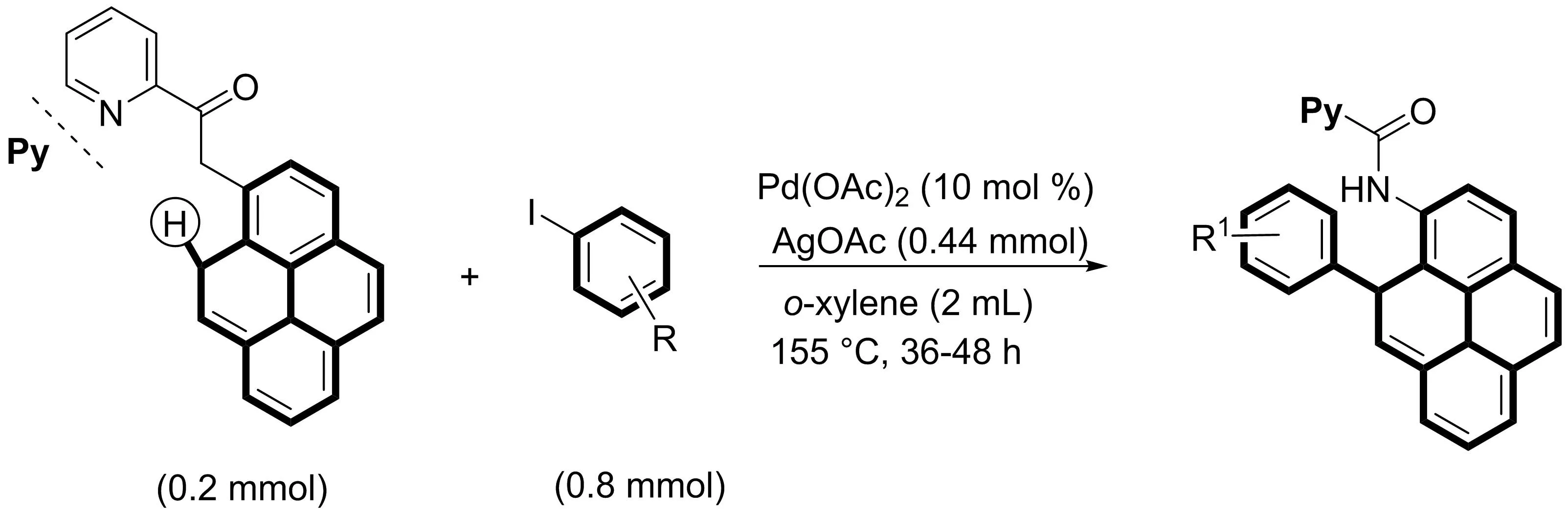
圖12 Pd(II)催化雙導向基團芘核的C-H芳基化和烷基化
2 銅催化吡啶甲酰萘胺的C-8位選擇性C-H官能化反應(yīng)
2013年,Miura課題組[15]報道了一種銅催化的1-萘胺和1,3-(苯并)噁唑脫氫性交叉偶聯(lián)反應(yīng)(圖13).其成功的關(guān)鍵是引入了以吡啶酰胺為導向基的N,N-雙齒配位體系.該反應(yīng)在廉價的過渡金屬銅催化劑的作用下順利進行,高產(chǎn)率得到π延伸率高的聯(lián)雜芳烴化合物.
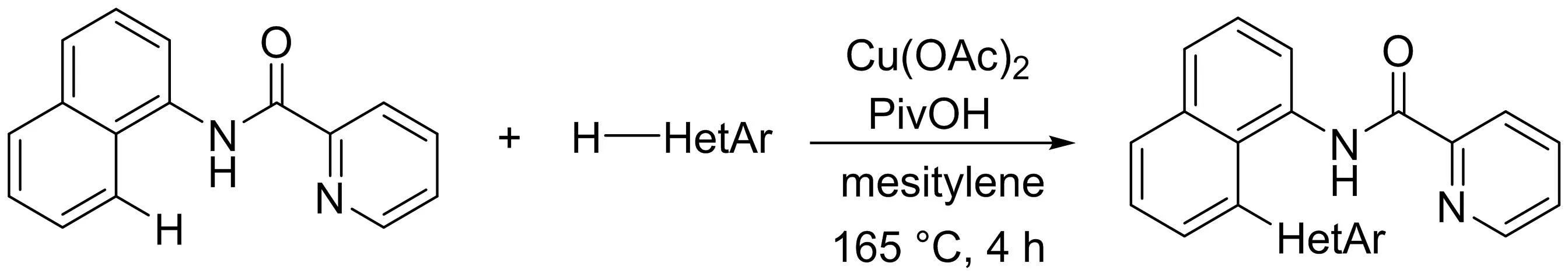
圖13 銅催化的1-萘胺和1,3-(苯并)噁唑脫氫二芳基偶聯(lián)反應(yīng)
孫志忠課題組[16]以苯甲酰甲腈為氰基源,發(fā)展了Cu(TFA)2催化下吡啶酰胺導向的1-萘胺衍生物C8-H直接氰化反應(yīng)(圖14).該方法可以以中等至良好的產(chǎn)率高效地得到一系列8-氰基-1-(吡啶酰胺)萘胺衍生物.苯甲酰氰化物首次用于芳烴的C(sp2)-H氰化反應(yīng),吡啶酰胺基團在萘的區(qū)域選擇性氰化反應(yīng)中起著重要的導向基團作用.
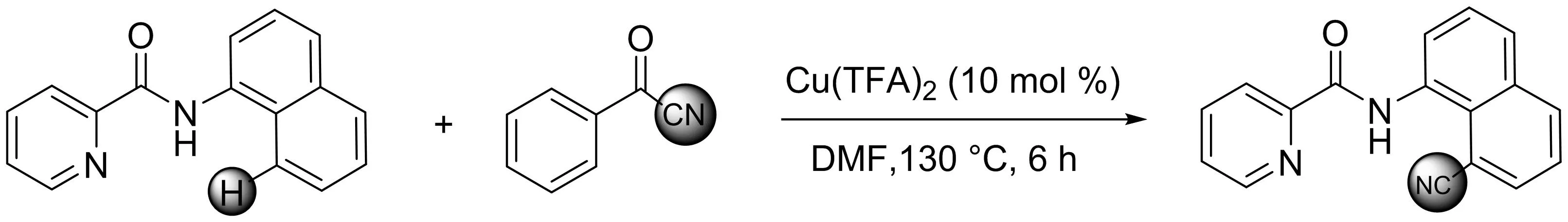
圖14 Cu(TFA)2催化吡啶酰胺導向的1-萘胺衍生物C8-H直接氰化反應(yīng)
Punniyamurthy課題組[17]以水為氧源,研究了銅介導的吡啶酰胺導向的1-萘胺與芳基硼酸的區(qū)域選擇性C-H官能化及C-O鍵的形成(圖15).動力學同位素研究表明,C-H鍵的活化是反應(yīng)速率的決定步驟.H218O標記實驗揭示了水中的氧插入到萘基和芳基之間.底物適用范圍廣、有良好的官能團多樣性和后期合成實用性是反應(yīng)重要的特點.
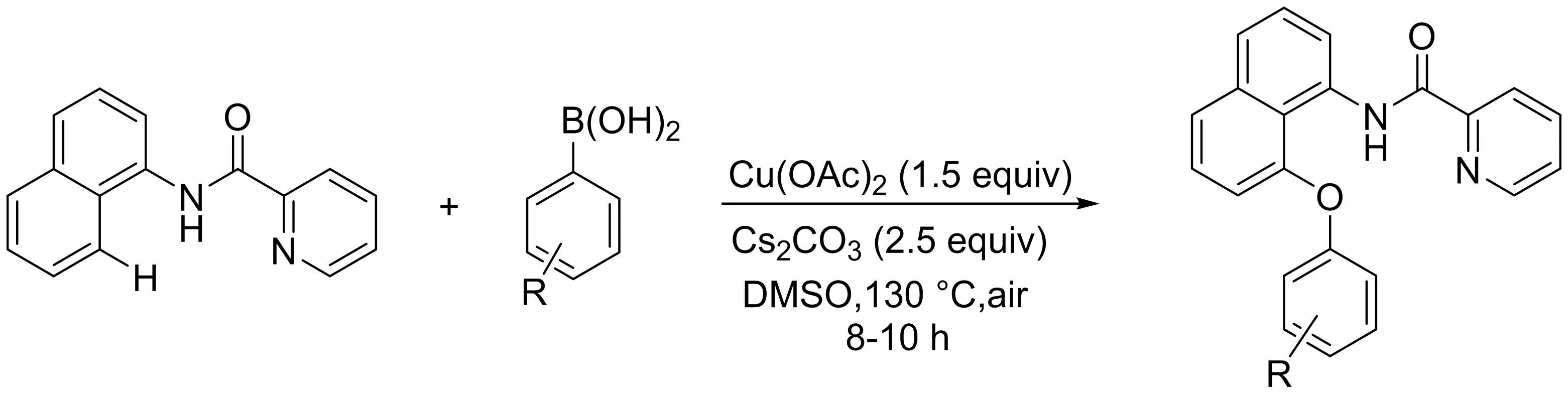
圖15 銅介導的萘酰胺與芳基硼酸的區(qū)域選擇性C-H醚化反應(yīng)
該課題組[18]隨后利用銅催化的吡啶酰胺導向的1-萘胺與二烷基丙二酸酯的氧化C-H/N-H環(huán)化反應(yīng)制備了有較高合成價值的二氫苯并吲哚酮化合物(圖16).該反應(yīng)通過C(sp2)-H/C(sp3)-H鍵直接活化,分子內(nèi)N-H/C(sp3)-H氧化脫氫偶聯(lián)來實現(xiàn)具有良好官能團耐受性的目標分子的合成.
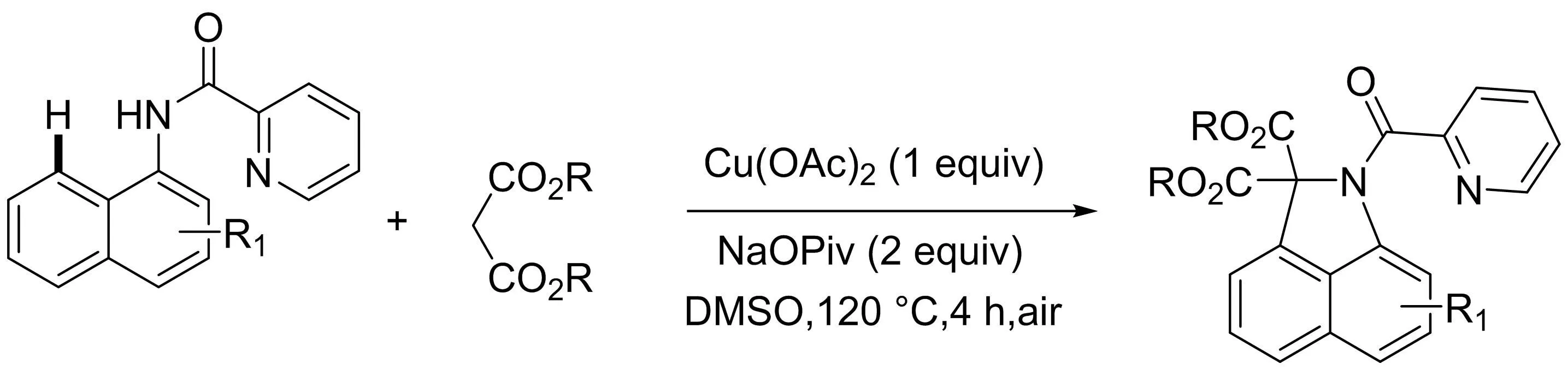
圖16 銅催化芳香酰胺與丙二酸二烷基酯的氧化C-H/N-H環(huán)化反應(yīng)
吳養(yǎng)潔課題組[19]以乙酸銅和過氧化二叔丁基(DTBP)為催化劑和氧化劑,發(fā)展了一種簡便高效的1-萘胺衍生物的C8-H直接胺化反應(yīng)(圖17).該反應(yīng)在無溶劑條件下即可順利進行,具有良好的區(qū)域選擇性和官能團耐受性,而吡啶酰胺在C-H官能化反應(yīng)中起著重要的導向基團作用.
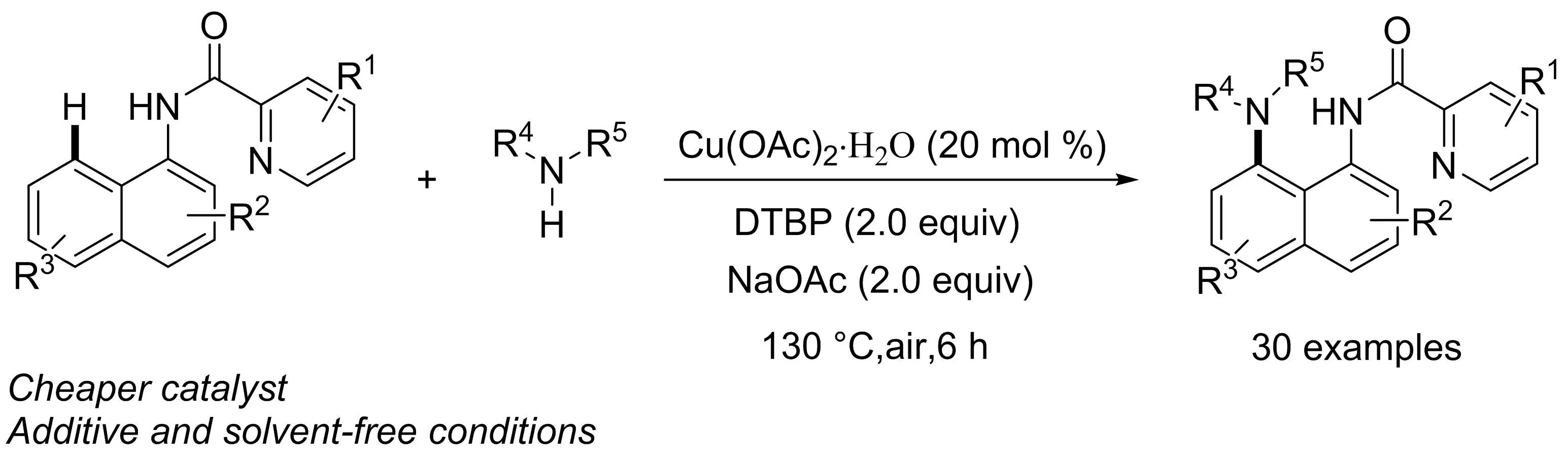
圖17 Cu(II)催化的1-萘胺在C8位的直接胺化反應(yīng)
最近Ghosh[20]也報道了一種類似的銅介導的1-萘胺衍生物C-8胺化反應(yīng).吡啶酰胺及其衍生物被用作C-8胺化反應(yīng)的雙齒導向基團(圖18).各種取代萘胺衍生物在溫和的條件下順利發(fā)生反應(yīng),而空氣作為氧化劑實現(xiàn)了脫氫性交叉偶聯(lián).
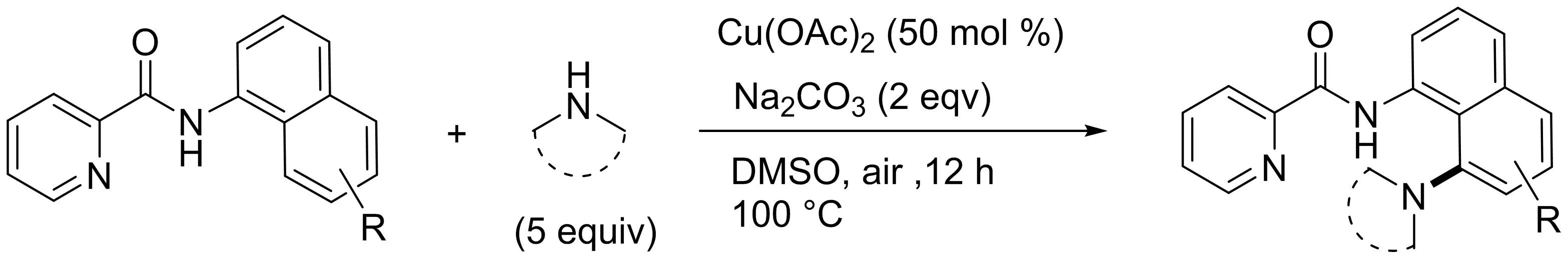
圖18 Cu(II)介導1-萘酰胺衍生物的C-8胺化反應(yīng)
3 鈷催化吡啶甲酰萘胺的C-8位選擇性C-H官能化反應(yīng)
Daugulis課題組[21]在2014年報道了一種鈷催化的吡啶酰胺導向C(sp2)-H鍵烯化反應(yīng)的方法(圖19).該反應(yīng)采用Co(OAc)2·4H2O催化劑,Mn(OAc)2共催化劑,內(nèi)炔烴作為烯基化試劑,底物氧氣作為末端氧化劑,產(chǎn)率中等.

圖19 鈷催化的吡啶酰胺導向的C(sp2)-H鍵炔烴烯基化反應(yīng)
吳養(yǎng)潔課題組[22]研究了鈷催化的1-萘胺衍生物與簡單易得的醇的區(qū)域選擇性C8-H鍵烷氧基化反應(yīng)(圖20),發(fā)展了一種制備具有良好官能團耐受性的8-烷氧基-1-萘胺衍生物的有效方法.值得注意的是,吡啶酰胺部分作為雙齒導向基團該區(qū)域選擇性轉(zhuǎn)化中發(fā)揮關(guān)鍵作用.
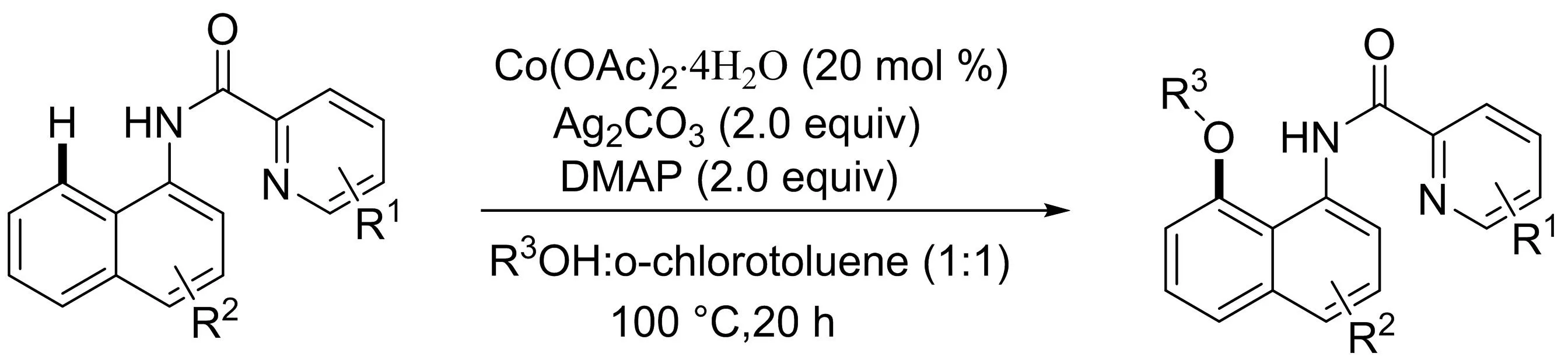
圖20 Co(II)催化1-萘胺衍生物與醇的C8-H烷氧基化反應(yīng)
隨后本課題組[23]發(fā)展了鈷催化1-萘胺與二醇的雙齒螯合區(qū)域選擇性C-H烷氧基化反應(yīng)(圖21).在該轉(zhuǎn)化過程中,直鏈二醇,支鏈二醇和聚乙二醇在現(xiàn)有反應(yīng)條件下都具有耐受性,從而生成相應(yīng)的端羥基烷氧基萘胺.此外,對照實驗表明,吡啶酰胺是C8-H烷氧基化反應(yīng)的關(guān)鍵導向基團,而且該反應(yīng)可能通過單電子轉(zhuǎn)移(SET)過程進行.
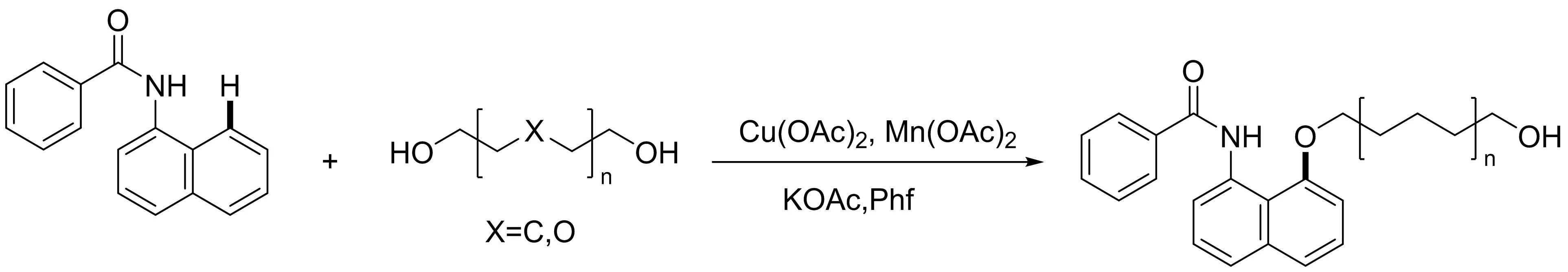
圖21 鈷催化1-萘胺衍生物與二醇的區(qū)域選擇性交叉脫氫C-O偶聯(lián)
吳曉峰課題組[24]報道了一種鈷催化的1-萘胺通過C-H鍵直接羰基化合成苯[cd]吲哚-2(1H)-酮骨架結(jié)構(gòu)的方法(圖22).反應(yīng)采用無痕導向基團,以苯-1,3,5-三甲酸三酯(TFBen)為CO源,得到各種苯[cd]吲哚-2(1H)-酮衍生物,產(chǎn)率中等至良好.作者還利用該方法實現(xiàn)了溴蛋白抑制劑BET的全合成.
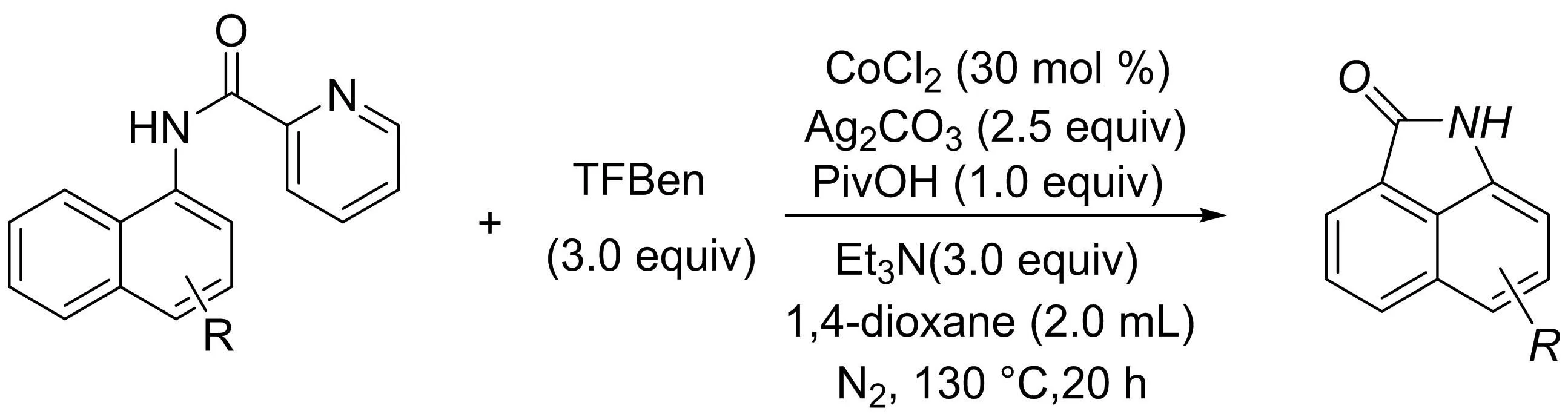
圖22 鈷催化1-萘胺直接羰基化合成苯[cd]吲哚-2(1H)-酮
吳養(yǎng)潔課題組[25]報道了一種簡便高效實現(xiàn)鈷催化的1-萘胺衍生物與芳基/烷基亞硫酸鈉的區(qū)域選擇性C8-H磺酰化反應(yīng)的方法,以中等至良好的產(chǎn)率得到的C-8選擇性磺酰化的萘胺衍生物(圖23).該反應(yīng)具有廣泛的官能團耐受性和較高的區(qū)域選擇性.需要注意的是,加入一定催化量的NFSI或氟試劑作為助劑可以提高反應(yīng)效率.此外,吡啶酰胺作為雙齒導向基團在這種區(qū)域選擇性轉(zhuǎn)化中發(fā)揮了關(guān)鍵作用.
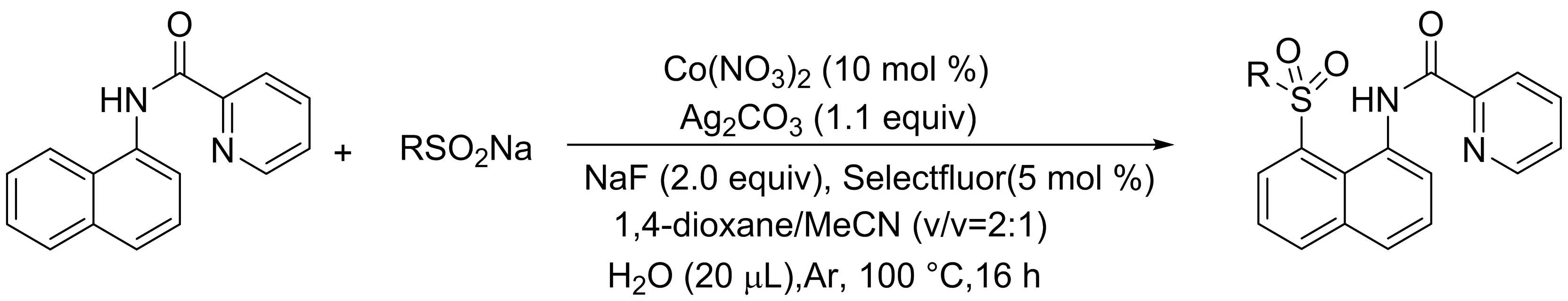
圖23 鈷催化1-萘胺衍生物與亞硫酸鈉的C8-H磺酰化反應(yīng)
王磊課題組[26]報道了一種基于鈷催化/光氧化還原的復合催化體系,實現(xiàn)了1-萘胺衍生物與醇的C8-H烷氧基化反應(yīng)(圖24).以市售的烷基醇為原料,Co(OAc)2和羅丹明B為催化劑,1-萘胺衍生物高產(chǎn)率的生成相應(yīng)的C8-H烷氧基化產(chǎn)物,官能團耐受性良好.
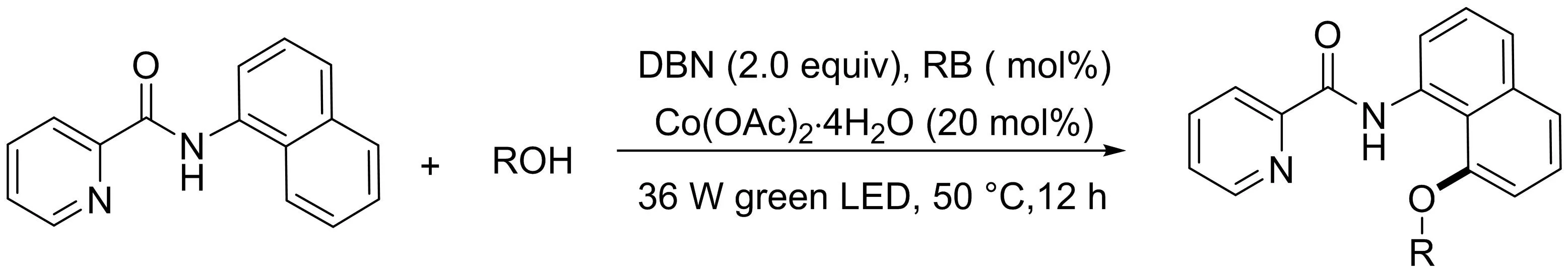
圖24 鈷催化1-萘胺衍生物與醇的C8-H烷氧
4 其他過渡金屬催化吡啶甲酰萘胺的C-8位選擇性C-H官能化反應(yīng)
Nakamura課題組[27]報道了在膦配體以及化學計量氧化劑的存在下的,三價鐵鹽催化下使用AlMe3作為甲基化試劑,可以實現(xiàn)C(sp2)-H鍵的C-8直接甲基化反應(yīng)的轉(zhuǎn)化(圖25).該反應(yīng)適用于含有吡啶導向基團的各種酰胺底物,使用溫和的鋁試劑可以防止鐵的還原,并且反應(yīng)的催化劑TON高達6 500.
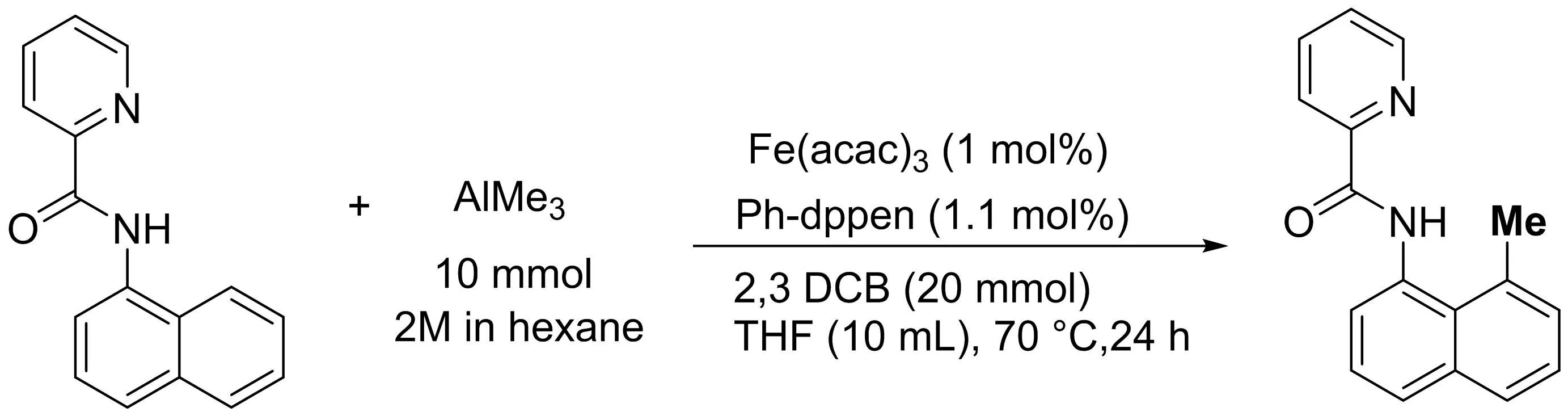
圖25 鐵催化三甲基鋁的定向C(sp2)-H和C(sp3)-H官能化
Chatani課題組[28]報道了銠催化的1-萘胺衍生物的C-H烷基化反應(yīng)(圖26).各種烯烴,包括苯乙烯衍生物,α、β-不飽和羰基化合物,甚至未活化的烯烴都可以作為偶聯(lián)底物.通過動力學研究、氘標記研究和控制實驗對反應(yīng)機理進行了研究,反應(yīng)可能是通過一種由烯烴生成的銠卡賓中間體進行的.
5 結(jié)論
綜上所述,在過去的十年里,過渡金屬催化下吡啶甲酰萘胺的雙齒導向的C-H鍵官能化反應(yīng)的領(lǐng)域迅速發(fā)展,涌現(xiàn)出各種類型的官能化轉(zhuǎn)化策略,并實現(xiàn)了各種碳-碳鍵或碳-雜鍵的構(gòu)建.文獻中報道了各種轉(zhuǎn)化,包括芳基化、烯烴化、烷基化、胺化、磺酰化、硫(硒)化等.大部分反應(yīng)由于其良好的官能團相容性和高反應(yīng)效率,已被廣泛用于功能分子的合成.
該領(lǐng)域仍然存在著進一步發(fā)展的空間,一方面導向基團不是產(chǎn)物的固有部分,需要安裝、移除或轉(zhuǎn)換為最終所需的官能團,另一方面,目前大多數(shù)C-H鍵的螯合導向金屬化僅限于鄰位選擇性或β-選擇性.控制C-H斷裂位置的選擇性,尤其是實現(xiàn)遠程C-H官能化,仍然是一個巨大的挑戰(zhàn).此外,不對稱C-H官能仍處于起步階段,還有很大的發(fā)展?jié)摿?為了解決這些問題,需要更加深入的機理研究,以全面了解C-H活化過程,總結(jié)催化劑、配體、溶劑、添加劑和底物之間復雜相互作用的規(guī)律,從而指導新的高活性和選擇性催化體系的設(shè)計和發(fā)現(xiàn).
