尿嘧啶激發態動力學溶劑效應的飛秒瞬態吸收光譜研究*
2022-09-30 05:41:56沈環華林強魏政榮
物理學報 2022年18期
沈環 華林強 魏政榮
1) (華中農業大學理學院,武漢 430070)
2) (中國科學院精密測量科學與技術創新研究院,波譜與原子分子物理國家重點實驗室,武漢 430071)
3) (湖北大學物理與電子科學學院,武漢 430062)
尿嘧啶是構成RNA 鏈最基本的結構單元之一,其光輻射下的光物理和光化學過程與光致癌變等過程緊密關聯,因而受到廣泛的關注.本文采用飛秒瞬態吸收光譜技術,研究了在紫外光輻射下,溶劑效應對尿嘧啶激發態超快動力學的影響.實驗利用一束264 nm 的激發光將尿嘧啶激發到1(π,π*)態(即S2 態).然后,利用覆蓋范圍為280—360 nm 的紫外超連續譜作為探測光,實現對尿嘧啶激發態和基態動力學的同步探測.測量發現,尿嘧啶在乙腈溶劑中顯現了兩個動力學衰減過程,即9.8 ps (在300 nm 處)和>1000 ps,分別對應于振動冷卻過程和三重態的衰減過程.這一觀察與水溶液中的結果差異很大,因為在水溶液中基本沒有測量到三重態的布居.通過對一系列溶液展開測量,發現尿嘧啶的超快動力學過程依賴于溶劑形成氫健能力的大小.越容易形成氫鍵的溶劑,尿嘧啶越不容易布居到三重態.
1 引言
核酸堿基是DNA 和RNA 的基本組成部分,是核酸分子中對光反應最敏感的部分.對核酸堿基在紫外光輻射下的光物理和光化學過程的理解,是研究DNA 光誘變損傷和光致癌的基礎[1-3],特別是光輻射下核酸堿基的超快動力學過程.因此,該方面研究的展開,不僅在基礎研究中具有重要的科學意義,還在生命健康方面具有重要應用價值.
尿嘧啶(U)是最簡單的天然核酸堿基.一般認為,在溶液中的尿嘧啶受到260 nm 左右的紫外光照射下被泵浦到激發態,隨后在激發態經歷內轉換過程衰變到基態,這一過程大概在皮秒量級.因此,盡管紫外光在自然界中普遍存在,但是在紫外光誘導下處于激發態的核酸堿基仍然能夠快速地回到穩定的基態.這一過程能夠使DNA 避免光致損傷并保持其遺傳信息[4-8].然而,最近新的研究表明,溶劑效應能夠對尿嘧啶以及衍生物的超快衰變動力學過程產生很大的影響[9-17].Hare 等[9]利用飛秒瞬態吸收光譜技術研究了1-環己基尿嘧啶(1-CHU)在質子和非質子溶液中的超快衰變動力學過程.研究發現,在質子溶劑中1-環己基尿嘧啶暗態1(n,π*)的非輻射弛豫時間尺度在幾十皮秒,而在非質子溶劑中,其弛豫時間尺度為數十納秒.他們提出假設,認為質子溶劑能夠調節明態1(π,π*)和暗態1(n,π*)的能級.當暗態1(n,π*)的能級和三重態3(π,π*)態的能級相近時,這兩個態之間就會發生系間交叉過程.Gustavsson 等[10,11]通過飛秒熒光上轉換光譜法測量了5-氟尿嘧啶(5-FU)在水、乙腈和甲醇中的激發態動力學.結果表明,溶劑對明態1(π,π*)的衰變動力學過程有顯著影響.這主要是與溶劑的氫鍵有關,而與溶劑的極性和黏度關系不大.在之前的研究中,我們報道了尿嘧啶及其C5,C6 取代衍生物在水和乙腈溶液中的飛秒瞬態吸收光譜研究,發現這些分子被泵浦到明態1(π,π*)之后,平行衰減到基態S0和中間態1(n,π*).并且,C5和C6 取代延緩了明態1(π,π*)到中間態1(n,π*)的衰減速率[12,13].最近,Li 等[14]采用共振拉曼光譜方法結合理論計算研究了6-氮尿嘧啶(6-AU)S2態的超快動力學過程,他們對S2態主要衰減至S1態的結論與我們瞬態吸收光譜測量的結果一致.Li 等的研究還就溶劑效應對尿嘧啶、6-氮尿嘧啶等體系超快動力學的影響展開了討論.在理論計算方面,Improta 等[15-17]利用可極化連續模型(PCM)研究了尿嘧啶及其衍生物的溶劑效應,在分子尺度上計算了5-FU 和U 在水和乙腈溶劑中超快衰變的溶劑效應.他們認為這種超快衰變過程有極大可能是受到溶劑與溶質分子之間氫鍵的強烈影響,因為5-FU 的1(π,π*)態和1(n,π*)態之間的錐形交叉過程將使C(4)=O 在乙腈溶劑中的鍵長拉長[14].Etinski 等[18]通過量子化學計算發現,6-氮雜尿嘧啶從1(n,π*)到三重態T1的超快系間交叉(intersystem crossing,ISC)過程在真空中為125 ps,而在乙腈溶液中為34 ps.他們的研究認為這種衰變壽命的差異主要受溶劑極性的影響.最近,Danillo 等[19]采用CASPT2 方法和cc-pVDZ 基組,計算了1-環己基尿嘧啶在真空以及水溶液中的超快動力學,并對極性質子型溶劑中三重態產率的抑制現象提出了解釋.
如上所述,盡管人們對尿嘧啶及其衍生物的衰變動力學過程做了很多研究,但是溶劑效應對其衰變過程的影響仍未得到全面理解.尤其是在紫外光輻射下的基態和三重態的動力學過程,由于瞬態吸收信號常常出現在紫外波段甚至深紫外波段,一直未能被直接觀察和深入理解.然而,這一困難可以通過使用延伸到紫外區域的連續性白光作為探測光來克服.在之前的研究中,我們將探測光延伸到300 nm 以下,這使得能夠同時研究尿嘧啶及其衍生物的基態和激發態的衰變動力學過程,從而能夠全面揭示取代基對尿嘧啶光輻射過程的影響[12,13].本文采用飛秒瞬態吸收光譜技術,在紫外區域對處于激發態的尿嘧啶在不同溶劑中的超快衰變動力學過程進行了研究.系統討論了溶劑環境對尿嘧啶的瞬態吸收光譜和激發態弛豫動力學過程的影響.研究發現,尿嘧啶三重態的布居動力學與溶劑的特征緊密相關.
2 實驗設備和過程
實驗用到的尿嘧啶樣品,采購于Alfa Aesar公司.實驗用到的溶劑采購于申試化工,為高效液相色譜(HPLC)級.所有的化合物在使用前未經過進一步提純.所有的實驗溶液均在實驗室中進行重新配制,從而保證實驗樣品的可靠性.所有測量均在室溫下飽和空氣條件下進行,樣品溶液的濃度為1 mmol/L.
在之前的研究中,我們詳細描述過實驗設備[12,13].這里進行一個簡要的描述.實驗中使用的飛秒激光器是摻鈦藍寶石多通放大器,型號為FemtoPower Compact PRO (Femtolasers Produktions GmbH),中心波長在792 nm,脈沖寬度30 fs,脈沖強度0.8 mJ,重復頻率5 kHz.激光器輸出的基頻光束經過分束器被分成兩束.一束基頻脈沖經過二倍頻晶體BBO 被轉換成396 nm 的二次諧波,然后和基頻脈沖進行合頻產生264 nm 的三次諧波(約2 μJ).這一束脈沖(264 nm)用來激發研究樣品到它的激發態1(π,π*)態,即S2態.將產生的396 nm的二次諧波的一小部分(約1 μJ)聚焦在CaF2晶體上用來產生探測光和參考光.產生的連續性白光波長可延伸到深紫外區域,波長覆蓋范圍為250—600 nm 可見光區域.樣品放置在1 mm 厚的流動循環樣品池中,以保證激光脈沖每次都與新樣品相互作用.通過調節泵浦脈沖和探測脈沖之間的時間延遲,使得這兩束脈沖在樣品上實現時間和空間上的重合.兩束激光脈沖均為線性偏振,它們之間的偏振夾角被設置為魔角(約54°),以消除分子取向效應帶來的影響(盡管實驗中并未發現激光偏振角度對實驗結果的明顯影響).兩個線性CCD 被用來測量探測光和參考光的光譜信號.實驗中使用的激光脈沖的波長均由光譜儀直接測量,而光譜儀則通過已知原子譜線進行校準.根據光學克爾效應,校正了作為探測脈沖的連續性白光對時間零點的影響,整個裝置的時間分辨率可達到300 fs 左右.實驗中,對數據進行5—10 次的重復采集,然后對平均結果進行處理.
3 實驗測量結果及分析
3.1 尿嘧啶在乙腈溶劑中的激發態動力學
實驗中選擇乙腈作為一種典型的非質子溶劑,用來研究尿嘧啶在該類溶劑中的激發態動力學.尿嘧啶在乙腈溶劑中的穩態吸收譜已經有詳細的報道[4,6],本次實驗沒有再次進行測量.實驗首先直接測量了在不同的泵浦和探測脈沖的時間延遲下尿嘧啶在乙腈溶劑中的瞬態吸收光譜,結果如圖1(a)和圖1(b)所示.圖1(a)給出了在較小的時間延遲1—19.5 ps 之間的光譜,而圖1(b)則給出了在較大的時間延遲59.5—79.5 ps 之間的光譜.從圖1(a)可以看出,隨著泵浦光的激發,尿嘧啶被瞬間激發到一個激發態上,該激發態有一個很寬的正吸收帶,波長范圍覆蓋280—310 nm,吸收峰值約在300 nm.在時間延遲從1—9.5 ps 之間,該吸收帶一直存在,但在時間尺度上展現了一個快速的衰減,衰減壽命約為10 ps,并在吸收峰值上出現了藍移現象,吸收峰一直往波長更小的方向移動,其光譜變化趨勢如圖1(a)中的箭頭所示.此外可以發現,瞬態吸收光譜信號在長波范圍內(如>310 nm)較弱且衰減較快.這些特征和振動冷卻的特性十分相似.從圖1(b)可以看出,在時間延遲59.5—79.5 ps 之間,瞬態吸收光譜信號并沒有明顯的變化.在280—340 nm 之間吸收信號僅觀察到非常微小的衰減.在大于340 nm 的范圍內吸收信號幾乎沒有變化,并在79.5 ps 時仍然觀察到平穩的吸收信號強度,這說明此時尿嘧啶分子依舊處于激發態.

圖1 在不同的泵浦和探測脈沖的時間延遲下尿嘧啶在乙腈溶劑中的瞬態吸收光譜 (a) 1—19.5 ps;(b) 59.5—79.5 psFig.1.Transient absorbance change of uracil in acetonitrile at different pump-probe delays: (a) 1—19.5 ps;(b) 59.5—79.5 ps.
為了進一步提取尿嘧啶分子激發態的詳細動力學信息,對瞬態吸收光譜中較慢的弛豫過程(>10 ps)進行了全局擬合分析,而對較快的過程在不同波長處進行了壽命的擬合.為了方便展示其主要特征,將300 nm 處的瞬態吸收光譜信號強度隨著泵浦-探測脈沖時間延遲的變化曲線,及其最佳擬合曲線均作圖于圖2 中.從圖2 可以看出,除了時間零點附近溶劑產生的背景信號,時間延遲變化曲線由一個快速衰減組分和一個慢速衰減組分組成.經擬合,快速衰減成分的壽命常數為9.8 ps,而慢速衰減成分的壽命常數>1000 ps.這一觀察結果與尿嘧啶在水溶劑中的情況差異較大.在水溶劑中觀察到的時間延遲曲線也有兩個衰減成份,但是衰減壽命常數分別為1.1 和22 ps,且1.1 ps 的快速衰減對應于基態S0的振動冷卻過程,而22 ps 則對應于最低的激發態1(n,π*),即S1態的壽命[13].這一結果表明在水溶劑中很難觀察到長壽命的系間交叉動力學過程,因為相當大部分的分子在1.1 ps 內就弛豫到了基態.在另一方面,早前的吸收光譜實驗報道了水溶劑中尿嘧啶的三重態T1的量子產率大約為0.02—0.04,而在乙腈溶劑中則有0.2[20,21].這意味著在乙腈溶劑中尿嘧啶的三重態T1的量子產率不能被忽略,并且在瞬態吸收光譜中有可能觀察到其長壽命的吸收信號.在本次測量中發現,在泵浦-探測時間延遲79.5 ps 之后尿嘧啶分子仍然處于激發態,并且顯示了一個非常緩慢的衰減趨勢(數據擬合壽命大于1000 ps).因此,我們認為該長壽命的衰減組分可以被標識為其三重態T1的壽命.

圖2 在300 nm 處尿嘧啶在乙腈中的瞬態吸收信號在不同的泵浦-探測光時間延遲下的變化曲線Fig.2.Time-dependent absorbance change of uracil in acetonitrile with different pump-probe delays at 300 nm.
在圖2 中,除了長壽命的組分,還有一個快速衰減的壽命組分,其時間常數為9.8 ps.這一結果與先前在非水溶劑中的分子基態振動冷卻的研究結果類似[22-26].此外,在圖1(a)中還觀察到了瞬態吸收峰的藍移現象,這也是基態振動冷卻過程中經常觀察到的一個典型特征.因此,我們將該組分歸因于基態(即S0態)的振動冷卻.此外,在探測光波長為295,310,325 和340 nm 處的擬合壽命分別為11,6.9,4.9 和1.7 ps.該壽命隨著波長的增加而減少.盡管如此,該壽命仍然要比在類似條件下水溶劑中的壽命要長得多.
本文無法明確分辨最低的激發態1(n,π*)態(即S1態)的吸收.在尿嘧啶分子中,其1(n,π*)態通常被認為是一種過渡態,通過它與T1態的系間交叉過程,可以實現三重激發態的布居[9].這與在水溶劑中不同,在水溶劑中可以清楚地觀察到壽命為22 ps 的組分,該組分被認為是1(n,π*)態的壽命[13].然而,我們在乙腈溶劑中并沒有清楚地觀察到與該壽命相近的組分,但是這不能排除1(n,π*)態被布居的可能性.這可能是因為在乙腈溶劑中尿嘧啶的1(n,π*)態吸收非常地弱,或者它與處于基態的高振動態相疊加,其動力學過程被混合進了基態的振動冷卻過程中.
基于以上分析,圖3 給出了尿嘧啶在乙腈溶劑中的衰減動力學過程示意圖.在264 nm 的激光脈沖激發后,分子被泵浦到1(π,π*)態.處于1(π,π*)的分子通過內轉換過程分別同時衰減到1(n,π*)和基態S0的高振動態.隨后,一部分分子通過1(n,π*)態衰減到三重態T1態,另一部分處于基態的高振動態的分子則進行振動冷卻的弛豫衰減,兩個過程對應的時間常數分別為>1000 ps 和9.8 ps.

圖3 尿嘧啶在乙腈溶劑中的光激發衰減動力學過程示意圖Fig.3.Jablonski diagram of the proposed decay mechanism for photo-excited uracil in acetonitrile.
3.2 尿嘧啶在甲醇溶劑中的激發態動力學
與乙腈溶劑不同,甲醇溶劑是典型的質子溶劑,我們也測量了尿嘧啶在甲醇溶劑中的超快激發態動力學.圖4(a)和圖4(b)給出了在不同的泵浦-探測光延遲下尿嘧啶在甲醇中的瞬態吸收光譜.與乙腈的情況類似,該瞬態吸收信號在280—350 nm區域顯示了快速衰減趨勢,壽命約為10 ps.此外,在最初的5 ps 內,吸收峰峰值在約290 nm 處,并且也觀察到了峰值明顯藍移的現象.同樣地,在20 ps 之后吸收光譜在290—350 nm 區域仍然顯示出很強的吸收信號,如圖4(b)所示.探測光在300 nm 處的瞬態吸收信號隨泵浦-探測光延遲的變化曲線也可以被擬合為兩個時間常數,即3.5 ps和>1000 ps.由于在乙腈和甲醇溶劑中觀察到的瞬態吸收光譜變化規律相似,我們將第一個快速時間常數標識為基態的高振動態的振動冷卻過程,而第二個慢速時間常數標識為三重態T1的壽命.
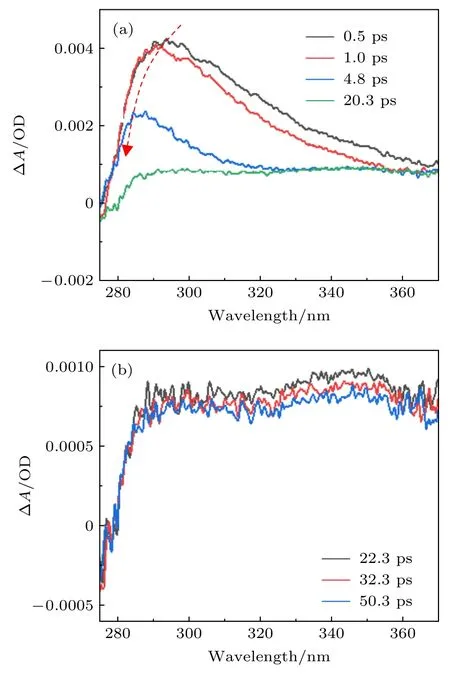
圖4 在不同的泵浦和探測脈沖的時間延遲下尿嘧啶在甲醇溶劑中的瞬態吸收光譜 (a) 0.5—20.3 ps;(b) 22.3—50.3 psFig.4.Transient absorbance change of uracil in methanol at different pump-probe delays: (a) 0.5—20.3 ps;(b) 22.3—50.3 ps.
3.3 尿嘧啶激發態動力學的溶劑效應
為了更加系統地研究溶劑對尿嘧啶激發態衰減動力學的影響,我們研究了水、乙腈、甲醇、乙二醇和乙酸乙酯等多種溶劑中的超快動力學(為了論文的可讀性,乙二醇和乙酸乙酯溶液的實驗數據未詳細顯示在本文中,水溶液的數據詳見參考文獻[12,13]).在這些溶劑中,乙腈和乙酸乙酯為非質子溶劑,其他則為質子溶劑,通過比較尿嘧啶在這兩類溶劑中的動力學過程可以揭示氫鍵的作用.此外,這些溶劑的黏度和極性也不相同.因此,這些溶劑的選擇可以更好地幫助我們系統地研究溶劑在尿嘧啶激發態衰減動力學過程中所扮演的角色.
通過比較發現,除了水溶劑之外,在其他大多數溶劑中都可以觀察到三重態的布居.如果用三重態衰減的振幅和振動冷卻過程來計算三重態的相對產率Y,顯示出以下趨勢:Y(乙腈) &Y(乙酸乙酯)>Y(甲醇) &Y(乙二醇)>Y(水).這一趨勢表明,在氫鍵較強的溶劑中,尿嘧啶以三重態存在的概率較低.還可以注意到,基態的振動冷卻過程的壽命顯示出類似的趨勢,即τ(乙腈) &τ(乙酸乙酯)>τ(甲醇) &τ(乙二醇)>τ(水),如表1 所列.這一趨勢表明,氫鍵加快了振動冷卻過程中的能量耗散,從而影響了三重態的產率.另一方面,還發現溶劑的黏度和極性對尿嘧啶的衰減過程的作用幾乎可以忽略不計.本文的研究結果與Hare 等[9]的結論具有一致性,即三重態主要來自1(n,π*)態,并且只有當1(n,π*)態具有多余的振動能量時,才能通過系間交叉過程過渡到三重態.本文的觀察結果表明,三重態T1被布居的前幾個皮秒內,在非質子溶劑中的振動冷卻過程很慢(通常在幾個或者幾十個皮秒內),這使得更多激發態的分子從熱態1(n,π*)過渡到三重態T1.
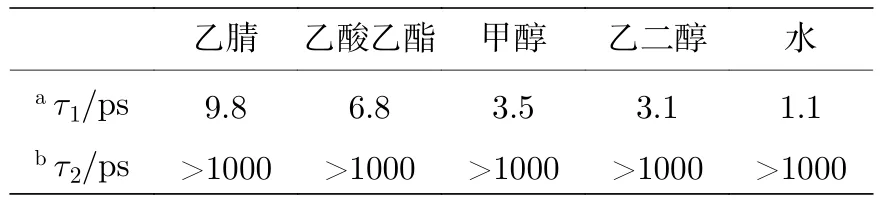
表1 在不同溶劑中觀察到的時間常數Table 1.Observed time constants in different solvents.
4 結論
本文利用飛秒瞬態吸收光譜技術,研究了尿嘧啶在不同溶劑中的激發態衰減動力學過程.實驗中使用了可延伸至280 nm 的深紫外探測光,更加直接地系統測量了尿嘧啶在不同溶劑中的三重態布居動力學.實驗結果為驗證三重態布居過程對溶劑的依賴性提供了直接有力的證據.通過對乙腈溶劑中測得的瞬態吸收光譜的詳細分析,在300 nm 處直接獲得了兩個衰減時間常數,即9.8 ps 和>1000 ps.與我們之前在水溶劑中沒有觀察到明顯的三重態的實驗結果相比,乙腈溶劑對三重態的布居起到了至關重要的作用.此外,系統測量了不同溶劑中尿嘧啶的瞬態吸收光譜,結果還表明尿嘧啶的衰減動力學對溶劑的氫鍵存在明顯的依賴性.在氫鍵較強的溶劑中,如甲醇、乙二醇和水溶劑中,尿嘧啶的1(n,π*)態分子轉移到三重態的概率較小且分子基態冷卻過程較快,這說明氫鍵加快了內轉換過程中的能量耗散速率.
感謝華心仲博士在早期實驗數據采集方面的貢獻.
