QuEChERS-氣相色譜—串聯質譜法測定茶葉中水胺硫磷農藥殘留的不確定度評定
2022-11-02 01:08:32陸振華于麗芳蔡丹萍
食品與機械 2022年10期
陸振華 于麗芳 蔡丹萍 李 成
水胺硫磷是由德國拜耳公司研發的一種廣譜性有機磷殺蟲、殺螨劑,具觸殺、殺卵和胃毒作用,在昆蟲體內能首先被氧化成毒性更大的水胺氧磷,抑制昆蟲體內的乙酰膽堿酯酶[1]。水胺硫磷屬于高毒農藥,已禁止在蔬菜、瓜果、茶葉、菌類、中草藥材上使用。GB 2763—2021《食品安全國家標準 食品中農藥最大殘留限量》中規定茶葉中水胺硫磷的最大殘留限量為0.05 mg/kg。近年來,茶葉中水胺硫磷超限量常有報道,如何提升農藥殘留檢測結果的準確性已成為相關檢測機構的一項重要工作。
目前,茶葉中農藥殘留的檢測方法較多。對氣相色譜法[2-4]、氣相色譜—質譜法[4-6]和液相色譜—質譜法[7]測定茶葉中農藥殘留的不確定度評定有較多報道,關于GB 23200.113—2018《食品安全國家標準 植物源性食品中208種農藥及其代謝物殘留量的測定 氣相色譜—質譜聯用法》測定蔬菜中農藥殘留的不確定度評定也有報道[8-9],而參照GB 23200.113—2018測定茶葉中農藥殘留的不確定度評定還未見相關報道。茶葉基質復雜,含有較多的生物堿、茶多酚和色素等對農藥殘留檢測產生干擾的物質,檢測時需加入更多的凈化試劑,且茶葉含水率較低,在提取過程中需用水浸泡使細胞膨脹,與有機溶劑充分接觸,才能達到提高提取效率的目的。且茶葉農殘檢測時,取樣量較少,稱樣量及均勻性對檢測結果的影響較大。因此茶葉的前處理對檢測結果的影響更大。對于各影響因素評定方法的處理,通常回收率有顯著性差異時,會將回收率代入計算公式以修正樣品的測定結果[10-11],但當實際出具的檢測數據滿足標準要求時,對回收率的處理并不直接折算到結果中。評定用內標法定量時的不確定度,通常的處理方法是用內標濃度進行評定再進行合成[12-13],但內標法一般用同一種內標,只與加標的體積有關。
研究基于CNAS-GL006—2019《化學分析中不確定度的評估指南》和JJF 1059.1—2012《測量不確定度評定與表示》,參照GB 23200.113—2018中QuEChERS-氣相色譜—串聯質譜法測定茶葉農藥殘留的方法,用單獨的數學模型將內標參與到評定過程中,將回收率以純度的方式參與到評定過程中,再對樣品稱量、體積引入、測量重復性等因素進行評定,找出影響茶葉中水胺硫磷檢測結果測量不確定度的主要原因,以期為實驗室檢測茶葉中水胺硫磷時進一步提高檢測數據的可靠性提供參考。
1 材料與方法
1.1 儀器與試劑
氣相色譜質譜聯用儀:GCMS-TQ8040型,日本SHIMADZU公司;
氮吹儀:N-EVAP 111型,美國Organomation公司;
旋渦儀:XH-B型,江蘇康健醫療用品有限公司;
超聲儀:KH-500DE型,昆山禾創超聲儀器有限公司;
離心機:TG1850-WS型,上海盧湘儀離心機儀器有限公司;
電子天平:SL2002N型,上海民橋精密科學儀器有限公司;
乙腈、乙酸乙酯:色譜純,美國Fisher公司;
水胺硫磷(CAS:24353-61-5;相對擴展不確定度±3%):1 000.0 μg/mL,北京曼哈格生物科技有限公司;
環氧七氯B(CAS:1024-57-3;相對擴展不確定度±3%):100.0 μg/mL,北京曼哈格生物科技有限公司;
乙二胺-N-丙基硅烷化硅膠(PSA):50 μm,納譜分析技術(蘇州)有限公司;
十八烷基硅烷鍵合硅膠(C18):50 μm,納譜分析技術(蘇州)有限公司;
石墨化炭黑(GCB):100 μm,納譜分析技術(蘇州)有限公司。
1.2 試驗方法
1.2.1 試樣處理
(1) 提取:準確稱取2 g粉碎均勻的茶葉樣品,于50 mL 塑料離心管中,加10 mL水渦旋混勻,靜置30 min。加入15 mL乙腈—醋酸(V乙腈∶V醋酸=99∶1)溶液、6 g無水硫酸鎂、1.5 g醋酸鈉、1顆陶瓷均質子,渦旋1 min,超聲30 min,4 200 r/min離心5 min。
(2) QuEChERS凈化:吸取8 mL上清液到15 mL塑料離心管中(內含1 200 mg硫酸鎂、400 mg PSA、400 mg C18及200 mg GCB),渦旋混勻1 min。4 200 r/min離心5 min。
(3) 濃縮:準確吸取2 mL上清液至10 mL玻璃試管中,40 ℃水浴中氮吹至近干。加入20 μL的內標溶液(環氧七氯B 5 μg/mL),加入1 mL乙酸乙酯復溶,過0.22 μm 濾膜,用于GC-MS/MS測定,內標法定量。
1.2.2 儀器檢測條件
(1) 色譜條件:色譜柱為TG-5MS(30 m×0.25 mm×0.25 μm);流量1.69 mL/min;程序升溫條件為初溫50 ℃,保留1 min,以25 ℃/min升溫至125 ℃,再以10 ℃/min 升溫至300 ℃,保留6 min;進樣口溫度250 ℃;不分流進樣,進樣量體積1 μL。
(2) 質譜條件:EI源;電子轟擊源70 eV;傳輸線溫度250 ℃;離子源溫度230 ℃;溶劑延遲2 min;多反應監測(MRM),優化后的質譜參數見表1。
1.2.3 測量數學模型
(1)
式中:
ω——待測樣品中水胺硫磷的含量,mg/kg;
c——試樣中水胺硫磷的質量濃度,μg/mL;
v——總的樣品代表的試樣體積,mL;
V1——加入提取溶劑的體積,mL;
V2——移取上清液的體積,mL;
V3——最終樣品復溶體積,mL;
m——試樣稱樣量,g;
fmethod——方法校正因子。
1.2.4 不確定度分量的主要來源 根據試驗的測定,茶葉中水胺硫磷殘留量測定不確定度分量主要來源如圖1所示。

表1 水胺硫磷及環氧七氯B的質譜參數表Table 1 Mass spectrum parameters of isocarbophos and heptachlor epoxide B

圖1 不確定度來源Figure 1 Analysis diagram of the uncertainty source
2 水胺硫磷農藥殘留不確定度分量的評定
2.1 樣品稱量引入的相對標準不確定度urel(m)

2.2 體積引入的相對標準不確定度urel(v)
主要包含加入提取溶劑V1、移取上清液的體積V2、最終乙酸乙酯復溶的體積V3。
2.2.1 加入提取溶劑引入的相對標準不確定度urel(V1)
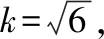
所以移液管移取15 mL乙腈—醋酸(V乙腈∶V醋酸=99∶1)溶液引入的相對標準不確定度為:
2.2.2 移取上清液引入的相對標準不確定度urel(V2)
移取2 mL上清液使用的是2 mL A級單標線吸量管,其允許誤差為±0.010 mL,參考15 mL單標線吸量管的評定過程,2 mL A級單標線吸量管引入的不確定度為:
實驗室溫度變化引入的標準不確定度為:
所以吸量管移取2 mL上清液引入的相對標準不確定度為:
2.2.3 乙酸乙酯復溶引入的相對標準不確定度urel(V3)
使用1 mL A級單標線吸量管移取1 mL乙酸乙酯復溶,1 mL A級單標線吸量管允許誤差為±0.007 mL,參考15 mL單標線吸量管的評定過程,1 mL A級單標線吸量管引入的不確定度為:
乙酸乙酯的體積膨脹系數為1.38×10-3℃-1,則溫度對乙酸乙酯體積變化引入的標準不確定度為:
所以移取1 mL乙酸乙酯復溶的相對標準不確定度為:
綜上,由提取液移取、提取液凈化分取和濃縮定容3個步驟引起的合成相對不確定度urel(v)為:
2.3 標準工作溶液校準過程引入的相對標準不確定度urel(c)
該相對標準不確定度主要來源:標準工作溶液的配制、標準工作曲線擬合。
2.3.1 標準工作溶液的配制引入的相對標準不確定度u1rel(c) 試驗使用的水胺硫磷標樣購于北京曼哈格生物科技有限公司,標準物質給出的質量濃度為1 000.0 μg/mL,其擴展不確定度為±3%(k=2),所以標準溶液的不確定度為:
10 μg/mL的標準儲備液的配制:使用量程為20~200 μL的移液器移取1 000.0 μg/mL的標準原液100 μL到10 mL容量瓶,用乙酸乙酯定容至刻度。
標準溶液稀釋過程:其數學模型為
(2)
式中:
cx——稀釋后標準溶液的質量濃度,μg/mL;
cs——標準原液的質量濃度,μg/mL;
vy——移液器移取的體積,μL;
vr——定容時容量瓶的體積,mL。

1 μg/mL儲備液:用1 mL A級單標線吸管吸取10 μg/mL 的儲備液1 mL到10 mL A級容量瓶中,用乙酸乙酯定容至刻度,其相對標準不確定度為:
標準工作溶液的配制:移取上述標準溶液1 mL到茶葉空白基質中,加20 μL環氧七氯B內標溶液(5 μg/mL),過膜,配制成質量濃度分別為0.005,0.01,0.05,0.1,0.5 μg/mL的標準工作溶液。1 mL標樣的移取使用的是1 mL A級單標線吸量管,樣品復溶時已有評定,這里不重復評定。試驗選擇與被測樣品接近的urel(c0.01)=2.223%予以評定,則u1rel(c)=urel(c0.01)=2.223%。
2.3.2 標準工作曲線擬合引入的不確定度 5種濃度的標準工作溶液由低到高分別上機進樣,得到的濃度比及面積比的數據見表2。采用儀器自帶的定量軟件最小二乘法擬合,得到標準工作曲線的回歸方程和相關系數為:
Arf=-0.025 042+5.544 347cri,R2=0.999 997,
(3)
式中:
cri——標準溶液與內標溶液濃度比值;
Arf——標準物質峰面積與內標峰面積的比值。
為了評定茶葉中水胺硫磷殘留限量時(即殘留量為0.05 mg/kg)的不確定度,所以假定某陽性樣品平行測定2次,得到樣液中平均質量濃度為0.013 33 μg/mL,內標質量濃度為0.1 μg/mL。其與內標濃度比的相對標準不確定度按式(4)計算:
(4)

表2 線性回歸方程擬合過程及結果?Table 2 Linear regression equation fitting and results
式中:
s——擬合標準工作曲線的標準偏差;
b——擬合標準工作曲線的斜率,b=5.544 347;
p——試樣測定的次數,p=2;
n——標準工作溶液的測定次數,n=5;
cr——試樣中水胺硫磷與內標濃度比的平均值,cr=0.133 3;

cri——標準工作溶液中水胺硫磷與內標的系列濃度比。



所以標準工作溶液校準引入的相對標準不確定度為:
2.4 重復性測量引入的不確定度urel(rep)

2.5 方法回收率引入的不確定度urel(R)
對市售茶葉按1.2試驗方法進行檢測,在水胺硫磷目標峰位置觀察其質譜圖,無法識別出水胺硫磷特征離子的樣品確定為陰性樣品。稱取6份陰性茶葉樣品,添加量為0.05 mg/kg,測定回收率,結果見表3。

表3 樣品添加回收率Table 3 Results of replicate recovery tests in samples
標準不確定度采用平均值的標準偏差計算得到:
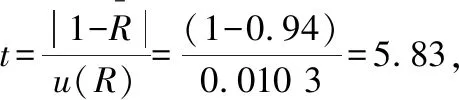
因此方法回收率引入的不確定度需進行修正。但通常情況下,檢測結果滿足標準要求時,不需要把回收率直接折算到檢測結果中,所以將回收率以水胺硫磷純度的方式參與到評定過程中,則
2.6 合成標準不確定度的評定
綜上所述,各因素相對不確定度分別為urel(m)=2.04%,urel(v)=0.778%,urel(c)=4.35%,urel(rep)=1.88%,urel(R)=3.37%,根據不確定度傳播公式對各分量進行合成:

u(ω)=0.050×6.22%=0.003 2 mg/kg。
2.7 擴展不確定度U(ω)
取包含因子k=2,置信水平P=95%,則擴展不確定度為:
U(ω)=2u(ω)=2×0.003 2=0.007 mg/kg。
測定結果:
通過QuEChERS-氣相色譜—串聯質譜法測定,該茶葉樣品中水胺硫磷殘留量結果為:
ω=(0.050±0.007) mg/kg,k=2。
3 結論
通過QuEChERS-氣相色譜—串聯質譜法測定茶葉中水胺硫磷殘留量的不確定度分析,得出該方法的檢測結果表示為ω=(0.050±0.007) mg/kg,k=2。從各標準不確定度分量數值大小可以看出,濃度c(涉及標準物質純度、標準儲備液與標準工作溶液的配制、標準工作曲線校準及內標加入量)引入的不確定度分量對合成標準不確定度貢獻最大,加標回收試驗引入的不確定度對合成標準不確定度的貢獻次之,因此,可以通過使用不確定度更小的標樣,用精度更高的量器配制標線及加內標等方式降低相關不確定度分量,也可以通過改善樣品的前處理步驟(QuEChERS凈化、氮吹等),減少水胺硫磷殘留量的損失,特殊情況,還可以通過回收率的校準,來提高結果的準確性,保證測量結果準確、可靠。
猜你喜歡
城市道橋與防洪(2022年4期)2022-07-01 06:04:12
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
當代陜西(2019年8期)2019-05-09 02:22:48
動漫星空(興趣百科)(2019年3期)2019-03-07 07:23:10
家庭影院技術(2018年4期)2018-05-09 07:07:52
海峽科技與產業(2016年3期)2016-05-17 04:32:12
